|
|
|
Indian Pediatr 2009;46: 425-427 |
 |
Genetic Studies in a Family with Distal Renal
Tubular Acidosis and Sensorineural Deafness |
|
Sidharth Kumar Sethi, Niranjan Singh, *Helena Gil
and Arvind Bagga
From the Division of Pediatric Nephrology, Department of
Pediatrics, All India Institute of Medical Sciences,
New Delhi, India and *Hospital Universitario Central de Asturias,
Asturias, Spain.
Correspondence to: Dr Arvind Bagga, Department of
Pediatrics, All India Institute of Medical Sciences, Ansari Nagar, New
Delhi 110 029, India. E-mail:
[email protected]
Manuscript received: March 18, 2008;
Review completed: April 7, 2008;
Accepted: May 14, 2008.
|
|
Abstract
Distal renal tubular acidosis (RTA) with
sensorineural deafness is a rare entity, inherited in an autosomal
recessive manner. It is caused by mutations in the ATP6V1B1 gene,
leading to defective function of H+-ATPase pump in the distal
nephron, cochlea and endolymphatic sac. We report two siblings with
distal RTA and sensorineural deafness having mutation C>T in the first
coding exon of the gene, resulting in a non functional protein. The
parents were found to be carriers for the mutation.
Key words: ATP6V1B1 gene, Renal tubular acidosis,
Sensorineural deafness.
|
|
Distal renal tubular acidosis (RTA) is
characterized by impaired urine acidification leading to severe
hyperchloremic metabolic acidosis, hypokalemia, hypercalciuria,
hypocitraturia, nephrocalcinosis and nephrolithiasis. The disease is
characterized by failure to thrive, nephrocalcinosis, polyuria and
urolithiasis. In untreated cases, the progression of nephrocalcinosis may
lead to chronic renal failure(1-3). Mutations in the ATP6V1B1 gene,
encoding the B1 subunit of vacuolar H+-ATPase, result in
autosomal recessive distal RTA associated with nerve deafness (OMIM
#267300). Genetic screening in multiple kindreds show different mutations
in the gene and almost all affected individuals have sensorineural hearing
loss. The majority of these mutations are likely to disrupt the structure
or abrogate the production, of the normal B1 subunit protein. This leads
to loss of expression of the gene in the human cochlea and in
endolymphatic sac epithelium, in addition to the renal tubular
defect(1,4).
We report two siblings with distal RTA and
sensorineural hearing loss having mutation in the first coding exon of the
ATP6V1B1 gene. Their parents showed the same mutation in a
heterozygous state.
Case Report
Patient 1. This 3-yr-old girl, born of
non-consanguineous marriage to a north-Indian Hindu family, presented with
complaints of polyuria, polydipsia, failure to thrive and bony
deformities. Similar history was present in an elder sibling who passed
away at 1-yr of age, but was not investigated. The parents were apparently
normal. Investigations showed metabolic acidosis (pH 7.08, bicarbonate 7.8
mEq/L) with plasma anion gap of 10 mEq/L; serum potassium levels ranged
between 2.7-3.2 mEq/L (Table I). Urine pH was
6.5, urine anion gap was positive and there was evidence of hypercalciuria
(urine calcium to creatinine ratio between 0.7-0.9 on multiple occasions).
Following bicarbonate loading, the fractional excretion of bicarbonate was
7.2% and difference between urinary to plasma CO 2
(U-P CO2)
was 1.7 mm Hg. Ultrasonography of abdomen showed bilateral medullary
nephrocalcinosis. Pure tone audiometry showed severe sensorineural nerve
deafness.
TABLE I
Biochemical and Genetic features*
| |
Patient 1 |
Patient 2 |
| Serum creatinine (mg/dL) |
0.4 |
0.7 |
| Sodium; potassium (mEq/L) |
138; 3.1 |
136; 3.1 |
| pH; bicarbonate (mEq/L) |
7.08; 7.8 |
7.16; 8.0 |
| Anion gap (mEq/L) |
10.0 |
11.4 |
| Calcium; phosphate (mg/dL) |
9.4; 3.2 |
9.5; 3.0 |
| Alkaline phosphatase (U/L) |
680 |
800 |
| Urine pH; anion gap (mEq/L) |
6.50; 11.2 |
6.12; 12.8 |
| Calcium/creatinine ratio (mg/mg) |
0.9 |
2.2 |
| Fractional excretion of bicarbonate |
7.2% |
6.8% |
| Urine - plasma pCO2 difference, (mm Hg) |
1.7 |
-4.6 |
| |
| Mutation in exon 1: C>T (R31X) |
Both siblings were homozygous, parents
heterozygous |
| * Both patients showed a severe growth
retardation with height and weight z-scores (WHO 2007)8 less than
-3, severe sensorineural deafness and nephrocalcinosis. |
Patient 2. This 1-year old younger sibling of
the first patient also presented with similar history of polyuria,
polydipsia, failure to thrive and bony deformities. Investigations showed
metabolic acidosis with plasma anion gap of 11.4 mEq/L; serum potassium
levels ranged between 3.0-3.2 mEq/L (Table I).
Urine pH was 6.12, urine anion gap was positive and there was evidence of
hypercalciuria (calcium to creatinine ratio ranging between 1.8-2.2 on
multiple occasions). Following bicarbonate loading, the fractional
excretion of bicarbonate was 6.8% and U-P CO 2
was -4.6 mm Hg. Ultrasonography of abdomen also showed bilateral medullary
nephrocalcinosis and audiometry showed severe sensorineural deafness.
A diagnosis of familial distal RTA with sensorineural
deafness was made. Both patients were treated with Polycitra-K to provide
8 mEq/kg/day bicarbonate, and were provided aural rehabilitation with
hearing aids and speech therapy.
ATP6VIBI gene sequencing: After informed
consent, DNA was extracted from the peripheral blood from the patients and
their parents. The 14 coding exons of ATP6V1B1 gene were amplified
by polymerase chain reaction using the primers described previously(1).
The amplified fragments were purified and sequenced on an automated ABI310
system, using BigDye chemistry (Applied Biosystems-Applera Corporaton,
Drive Foster City, CA). Sequencing of the coding exons and the intronic
flanking region showed DNA mutation at exon 1: C>T (R31X). Both siblings
were homozygous, while parents were heterozygous for the mutation (Fig.1).
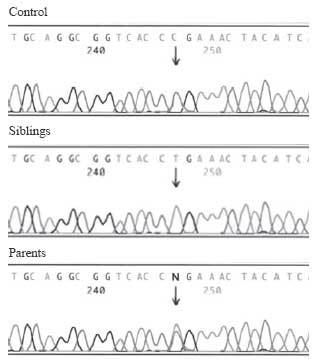 |
|
Fig.1 The electrophoregrams show
the exon 1 sequences of the ATP6V1B1 gene of a normal control and
the family. Both siblings were homozygous for the mutation exon 1:
91 C>T, and the both parents were carriers for the same mutation. |
Discussion
Both patients in the present report had features of
distal RTA with failure to thrive, polyuria, refractory rickets,
hypokalemia and nephrocalcinosis. They also showed severe sensorineural
deafness requiring rehabilitation. Investigations confirmed the diag-nosis
of secretory distal RTA and homozygous mutations were found in the
ATP6V1B1 gene in both cases.
Most cases of primary distal RTA in children result
from defective function of the proton pump vacuolar H +-ATPase,
located at the apical surface of the
a-intercalated
cells. The vacuolar H+-ATPase
is formed by several subunits. Mutations in the ATP6V0A4 gene,
which encodes for the a4 subunit, cause autosomal recessive dRTA (OMIM #
602722)(3-5). ATP6V1B1, a gene on chromosome 2, encodes for the
B1-subunit of the vacuolar H+-ATPase.
ATP6V1B1 is also expressed in the human cochlea and in
endolymphatic sac epithelium(4). Endolymph is a unique extracellular fluid
having low sodium and high potassium concentrations, which maximizes the
sensitivity of hair cells. To preserve its pH at 7.4, there is presumably
a requirement for proton pumping into endolymph. It is assumed that H+-ATPase
contributes to maintenance of endolymph pH and defects in its B1 subunit
cause irreversible hair cell damage because of abnormalities in
electrolyte and pH(6).
Apart from the presence or absence of hearing loss,
there do not appear to be major
phenotypic differences at diagnosis between patients with homozygous
ATP6V1B1 and ATP6V0A4 mutations. Long term follow-up of a
cohort of affected individuals with ATP6V0A4 mutations shows mild
and/or older-onset hearing impairment in some patients who were initially
considered to have normal hearing by audiometry(3).
Both siblings in this report were homozygous for a
known mutation in exon 1:C>T (R31X) in the ATP6V1B1 gene, as
previously reported by Karet, et al.(4). This change introduces a
termination codon at 31 position, resulting in a non functional
protein(4). Systemic alkali therapy, although correcting systemic pH,
fails to prevent progressive hearing loss in patients. It is important to
note that after 3 years of follow-up of our kindred, the sensorineural
hearing deficits still persist. Failure of alkali treatment to address the
abnormal endolymphatic physiology is believed to account for the
progressive hearing loss(4,7). Genetic evaluation of the affected family
has important implications for genetic counseling and understanding the
pathogenesis of the rare association.
Contributors: SKS and AB conceived and designed the
study and provided important intellectual content. NS drafted the paper
and helped in manuscript writing. HG conducted and interpreted mutational
analysis. The final manuscript was approved by all authors.
Funding: None.
Competing interests: None stated.
References
1. Gil H, Santos F, García E, Alvarez MV, Ordóñez FA,
Málaga S, et al. Distal RTA with nerve deafness: clinical spectrum
and mutational analysis in five children. Pediatr Nephrol 2007; 22:
825-828.
2. Laing CM, Toye AM, Capasso G, Unwin RJ. Renal
tubular acidosis: developments in our under-standing of the molecular
basis. Int J Biochem Cell Biol 2005; 37: 1151-1161.
3. Karet FE. Inherited distal renal tubular acidosis. J
Am Soc Nephrol 2002; 13: 2178-2184.
4. Karet FE, Finberg KE, Nelson RD, Nayir A, Mocan H,
Sanjad SA, et al. Mutations in the gene encoding B1 subunit of H +-ATPase
cause renal tubular acidosis with sensorineural deafness. Nat Genet 1999;
21: 84-90.
5. Stover EH, Borthwick KJ, Bavalia C, Eady N, Fritz
DM, Rungroj N, et al. Novel ATP6V1B1 and ATP6V0A4
mutations in autosomal recessive distal renal tubular acidosis with new
evidence for hearing loss. J Med Genet 2002; 39: 796-803.
6. Sterkers O, Saumon G, Tran Ba Huy P, Ferrary E,
Amiel C. Electrochemical heterogeneity of the cochlear endolymph: Effect
of acetazolamide. Am J Physiol 1984; 246: F47–53.
7. Rodriguez-Soriano J, Vallo A, Castillo G, Oliveros
R. Natural history of primary distal renal tubular acidosis treated since
infancy. J Pediatr 1982; 101: 669-676.
8. World Health Organisation. Child growth standards. Available from:
URL: http://www.who.int/nutrition/media_page/en/. Accessed on 30 April
2008.
|
|
|
 |
|
