|
|
Case Reports Indian Pediatrics 2006; 43:440-445 |
||
|
Berardinelli-Seip Congenital Lipodystrophy |
||
|
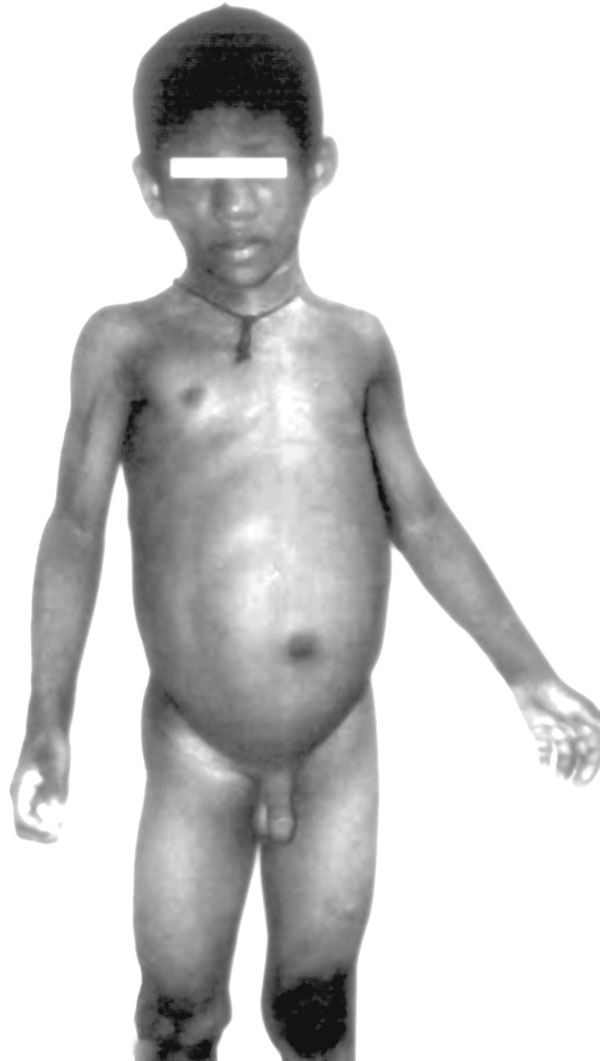
Abstract: Key words: Berardinelli-Seip congenital lipodystrophy, BSCL2 gene, renal anomaly. Berardinelli-Seip congenital lipodystrophy (BSCL) is a rare autosomal recessive disorder. Berardinelli from Brazil described the first patients in 1954. The syndrome was confirmed by Seip in Norway in 1959. It has been variously described as generalized lipodystrophy, congenital lipodystrophy, Seip-Lawrence syndrome and lipoatrophic diabetes (in USA). BSCL families are classified into BSCL1, BSCL2 and BSCLX(1). BSCL1, prevalent in African-American population, is the milder variety presenting in the second or third decade of life. Garg, et al.(2) first identified the gene for BSCL1 on chromosome 9q34. BSCL2 is more severe with onset in the neonatal period or early infancy. Most have mental retardation. It is prevalent in Portugal, Lebanon, Norway and Middle East. The locus for BSCL2 has been identified on chromo-some 11q13 by Magré, et al.(3). Worldwide; the prevalence of BSCL2 is somewhat more than that of BSCL1. BSCLX families are very rare; they show evidence against co-segregation with either 9q34 or 11q13. Agarwal, et al.(4) have detected different mutations of the gene (AGPAT2) encoding 1-acylglycerol-3-phosphate O-acyltransferase 2 in 20 affected individuals showing linkage to chromosome 9q34 (BSCL1). The AGPAT2 enzyme is involved in the biosynthesis of triacylglycerol and glycerophospholipids. In the 11q13 linked families (BSCL2) various homozygous and compound heterozygous mutations were found in a previously unidentified gene, termed seipin. More than 15 different allelic variations of the BSCL2 gene have been described(1,3). The seipin gene encodes for a 398-amino acid protein of unknown function. Magre, et al.(3) in 2001 indicated that this protein could be a transmembrane protein. Windpassinger, et al.(5) in 2004 showed that seipin is an integral membrane protein of the endoplasmic reticulum. This gene is widely expressed, with particularly high expression levels in brain and testis. In all BSCL subjects, there is near total absence of adipose tissue. Dietary and endogenous fat is aberrantly stored in metabolically important tissues like muscle and liver. This leads to severe insulin resistance and ultimately a difficult to control diabetes mellitus. Other metabolic consequences include hypertriglyceridemia leading to hepatic steatosis and ultimately to cirrhosis and death due hepatic failure. For unknown reasons many subjects develop hypertrophic cardiomyopathy that can lead to death from cardiac failure(1). Association with bone cyst has been reported; it was previously described as Brunzell syndrome. In this report we describe a newly identified allelic variant of the BSCL2 gene (found in family1) and its association with renal anomaly. Case Report Family 1 (case 1 and case 2) A pair of 3-year-old twins, came to our out patient department, with the presenting complaints of abdominal distension, abnormal facies, darkening of the skin and excessive hair all over the body since early infancy. The children are a product of non-consanguineous marriage. The mother had a history of two mid-trimester abortions prior to the birth of the twins. Antenatal period was uneventful except absence of one kidney in antenatal ultrasound in one child. Twins A had a height of 96 cm (between 50th and 75th percentiles of NCHS standard) and a weight of 15 kg (between 75th and 90th percentiles). Similarly, twins B showed a height of 95 cm (between 50th and 75th percentiles) and a weight 14 kg (between 50th and 75th percentiles). Both the twins have lack of subcutaneous fat in all body parts, apparent muscular hypertrophy, coarse and hyperpigmented skin, acanthosis nigricans in the nape of the neck, excessive body hairs (sparing the face, axilla and pubic area), hypertrophic genitalia and large superficial veins. The children had hepatomegaly and splenomegaly without features of cirrhosis or portal hypertension. Mental retardation was present in both twins. Laboratory investigations revealed normal hemogram, serum electrolytes and kidney function tests (urea and creatinine) in both. Liver enzymes were elevated (SGOT = 81 IU/L, SGPT = 143 IU/L, ALP = 1717 IU/L in twin A and SGOT = 95 IU/L, SGPT = 196 IU/L, ALP = 747 IU/L in twins B). Serum triglycerides was elevated (286 mg/dL in twins A and 284 mg/dL in twins B); serum total cholesterol was normal. Blood sugar was normal. Plasma insulin levels were elevated, suggesting insulin resistance [fasting = 32 µIU/mL and postprandial = 39 µIU/mL in twins A; and fasting = 30 µIU/mL and post-prandial = 58.5 µIU/mL in twins B; (normal = 2 to 25 µIU/mL)]. X-ray of skull, trunk and limbs has shown normal bones. Ultrasound of abdomen revealed enlargement of liver with increased echogenicity (suggestive of fatty liver) and enlarged spleen in both siblings. In twins A left kidney was absent in left renal fossa, while a cystic structure arising from the left posterolateral wall of the bladder, suggestive of ureterocoele was seen; right kidney was showing compensatory hyperplasia. ELISA for human immunodeficiency virus (HIV) was non-reactive and echocardiography was normal in both. In view of the typical dysmorphology, age of onset and hypertriglyceridemia, the diagnosis of BSCL2 was kept(6). Genetic studies were carried out. Both the children are coming for follow up since the last one-year. They are found to be prone to upper respiratory tract infections and skin abscesses, but respond well to oral antibiotics. As per recommendations the children are put on low fat diet (fat intake was restricted to 20 to 30%). Estimation of serum lipids and blood sugar is being done in six monthly intervals. None of the children have developed overt diabetes and serum triglycerides has somewhat decreased with dietary restriction. Special education for the children has been advised because of the associated mental retardation. The parents have been explained the frequency of recurrence of the disease and the importance of prenatal diagnosis in future pregnancy. Family 2 (case 3) From the old records of the Pediatric endocrine clinic a second family was detected. In July 2002 two male siblings, one-aged 3 years and the other 4 months, from this family (with history of consanguinity between parents), were evaluated for phallic hyper-trophy, but no cause could be found at that time. One of the siblings had expired at 4 month of age due to unknown cause before any evaluation could be done. Old records revealed hepatomegaly and hypertrichosis in both siblings along with phallic hypertrophy. Liver biopsy performed on the older sibling at that time, revealed fatty liver without any definite histological pattern. Estimation of 17 alpha hydroxy progesterone in three occasions in this child was normal. No cause for penile hypertrophy or hepatomegaly could be found at that time. The family was lost to follow up for almost two years. The 3-year male child was traced and investigated. He is now 6 years. He has typical dysmorphology suggestive of congenital lipoatrophy of BSCL2 type. He has lipoatrophy, acanthosis nigricans, hepatosplenomegaly and mental retardation (Fig. 1). He has deranged liver function test, hypertriglyceridemia (373 mg/dL) and insulin resistance (fasting 42 µIU/mL and postprandial 53 µIU/mL). ELISA for HIV is non-reactive. X-ray of bones and echocardio-graphy are normal. Ultrasound of abdomen revealed enlargement of liver with increased echogenicity, splenomegaly and normal kidneys.
The family is complete with two other alive and unaffected siblings. This child is also put on low fat diet, kept under follow up and advised about special education because of associated mental retardation. Genetic analysis of the affected children DNA of all the three subjects (cases 1, 2 and 3) was separated from their blood samples by "Trizol method". The packed cell fraction was subjected to RBC lysis by RBC lysis buffer. The separated WBC was treated with ‘Trizol reagent’ and centrifuged adding chloroform to separate RNA, DNA and protein. The interphase DNA was precipitated with ethanol and washed; the fresh DNA was dissolved in ‘TE buffer’. The DNA samples were sent to ‘Institute of Human Genetics’ in Belgium, for analysis. On analysis, mutations detected in the BSCL2 gene in the two families were different. In both the twins from family1, homozygosity for the c.IVS2-11 A>G has been found. Silico analysis suggested a splice site mutation. To confirm the hypothesis and to rule out a single polymorphism, blood was sent to the same institution and RNA study was carried out. The c.DNA (which is a copy of the mature spliced mRNA) analysis confirmed that this variant of BSCL2 gene creates the activation of a 3´ cryptic splice site. This abnormal splicing results in a 10 Bp insertion (followed by a frame shift) in exon 3. This is a newly identified allelic variant of the BSCL2 gene; this mutation of the BSCL2 gene has not been described earlier. Mutation detected in the BSCL2 gene in the child of family2 is homozygosity for the c.636delC or p.Tyr213Thr fs mutation (numbering with ATG = 1). This mutation has been described earlier in literature(1). The results confirmed the clinical diagnosis of BSCL and allowed assignment to type 2. Discussion We suspected generalized lipoatrophy from the appearance of the two siblings of the first family. The second family could be traced from old records by hind vision. The diagnosis was confirmed after the typical dysmorphology was noted and hypertri-glyceridemia and hyperinsulinemia were detected. The prevalence of BSCL is estimated to be 1 per 0.2 million in Lebanon, 1 per 0.5 million in Portugal, 1 per 1 million in Norway and 1 per 12 million in USA(6). Though some cases have been reported in Indian literature, genetic studies of those subjects were not carried out. The major diagnostic features of BSCL, described by Maldergem(6) are lipoatrophy, acromegaloid features, hepatomegaly, elevated serum triglycerides and insulin resistance (manifested as acanthosis nigricans); all of these are found in our patients. Other features include hypertrophic cardio-myopathy, mental retardation, hypertrichosis, bone cysts and precocious puberty (in females). All the three children were mentally retarded. Hypertrichosis was present in all at presentation. X-rays have not detected any bone cyst. Previously, lipoatrophy along with bone cyst was described as Brunzell syndrome. Later on it was found that bone cyst is only an association of BSCL(6). The skin changes like hypermelanosis, hypertrichosis and acanthosis nigricans found in our patients are similar to that found by Reddy, et al.(7) in 1986. These children can have associated immune deficiency as described by Kher, et al.(8) in 1990. This may be the cause of recurrent upper respiratory tract infections and skin infections in the affected twins of family 1. We have noted the association of renal anomaly (absence of one kidney) in twin A of the first family; this association was not reported earlier. Total lipoatrophy can be at times acquired (Lawrence syndrome) after an infectious illness and is presumed to be of autoimmune origin(9). Such associations are ruled out in our patients. Patients of Dunningan-Koberling(10) partial face sparing lipo-dystrophy have a distinctive cushingoid face, unlike total lipoatrophy, as found in our patients. In adults lipodystrophy can be at times found in association with HIV infection(11), especially as a side effect of anti-retroviral drugs. HIV infection has been ruled out in all the three children. Magre, et al. in 2001 determined that the BSCL2 gene contains 11 exons spanning at least 14 kb, with the putative initiation codon located in the second exon. Various allelic variants of the gene have been reported(3). The children from family1 (case 1 and 2) of our study, was found to be homozygous for c.IVS2-11 A >G [substitution of Adenine by Guanine (A >G) at position - 11 in intron 2 (IVS2-11) i.e., at position - 11 from exon 3, in their BSCL2 gene](12). The c.DNA analysis confirmed a splice site mutation as described previously. Though mutations in the introns (IVS) of the BSCL2 gene have been reported(3), this is a new variant. In the child from family 2 (case 3) in our study, the coding DNA reference sequence shows a deletion of a C at nucleotide 636. In the protein sequence, this causes a substitution of tyrosine (Tyr) by threonine (Thr) at position 213 (numbering with ATG = 1), thus causing a frame shift. They were homozygous for this mutation. Interestingly, Maldergem, et al.(1) have identified a similar mutation in the BSCL2 gene of a child of Indian origin, residing in United Kingdom. He was born in the year 1985, and diagnosed at 4 months of age; the cause for referral being organomegaly. His cardiac ultrasound was normal; and developed diabetes at the age of 15 years. He also had history of parental consanguinity. Genetic study is helpful in determination of the prognosis, since BSCL1 is a milder disease with lower occurrences of mental retardation and premature death than BSCL2. Recurrence risk is 25%, so chorionic villus sampling at 9 to 12 weeks is recommended for prenatal diagnosis in at risk families(6). We have made a clinical as well as a genetic diagnosis of BSCL2 in our patients. We report this new mutation (c.IVS2-11 A>G) detected in one of the families. We also report the association of renal anomaly with this mutation. Acknowledgements We acknowledge the contributions of Professor Lionel Van Maldergem, Head of the Department of Medical genetics and Professor Pascale Hilbert, Head of Mole-cular genetics, Institute de Pathologie et de Genetique, 41 Alle des Templiers, 6289 Gerpinnes, Belgium, Professor Madhulika Kabra, All India Institute of Medical Sciences, New Delhi, India, Dr. Anubhuti, Senior Resident, Department of Bio-chemistry, Lady Hardinge Medical College, New Delhi, India and Bintili Biswas, Junior Research Fellow, Depart-ment of Biochemistry, All India Institute of Medical Sciences, New Delhi, India. Contributors: KM and SA were involved in acquisition of data, drafting the article, revision and final approval of the version; AS and AK helped in acquisition of data, and final approval of the version;. Funding: None. Competing interests: None stated..
| ||
|
References | ||
|
|
![]()