|
|
Case Reports Indian Pediatrics 2003; 40:418-423 |
||||
|
Fowler-Bike Syndrome with Extreme Oligohydramnios, Growth Restriction and without Muscular Hypoplasia |
||||
|
Ashutosh Halder Inusha Panigrahi Lily Pal*
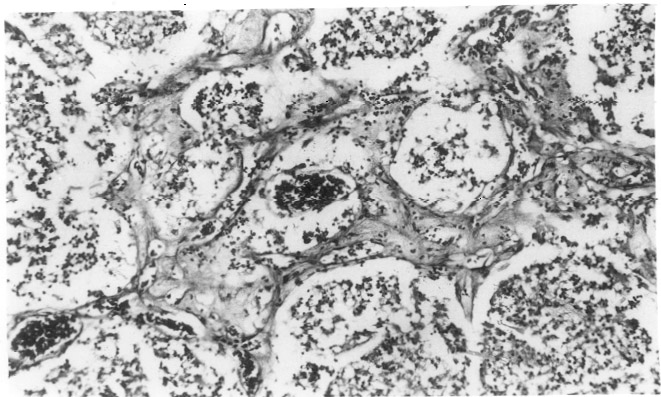
Fetal ventriculomegaly is a common malformation having varied etiology. How-ever, fetal ventriculomegaly in association with vascular proliferation and diffuse calcification in CNS has been reported in English literature only six time before(1-6). The present paper reports a similar occurrence in a family from Indian subcontinent. This case was slightly different from others requiring documentation with this syndrome. Case Report This male neonate was the product of first pregnancy of young, healthy, non-consangui-neous, Hindu Indian couple aged 22 (mother) and 27 (father) years. Pregnancy was under obstetric supervision from eighth week of pregnancy. Routine obstetric ultrasound at 16th week was normal. Pregnancy was uncomplicated until 29th week when second obstetric ultrasound was advised for clinical suspicion of growth restriction and oligo-hydramnios. Mother was normotensive and without any medical or pregnancy related problems. Second ultra-sound examination revealed mild ventriculo-megly in addition to growth restriction and oligohydramnios and was referred to our center for chromosomal analysis and counseling. Ultrasound examina-tion at our center confirmed above findings. Following counseling, couple refused to undergo invasive prenatal diagnostic proce-dure; and opted for ultrasound follow up and early delivery. Pregnancy was followed up at our center for next 6 weeks before induction of labour at 35th week. Follow-up ultrasound examination showed progressive oligo-hydramnios, growth restriction and ventri-culomegaly. Maternal serological studies (IgG & IgM) for rubella, cytomegalovirus and toxoplasma were negative for active infection. A male baby weighing 900 g was born at 35 weeks. The weight of placenta was 125 g. Apgar score at 5 minutes was 6/10. Movements of all four limbs were seen. Examination of baby revealed proptosis with corneal haziness on right eye, microphthalmia on left, depressed and pulsatile anterior fontanelle, clenched hands of both side and prominant heel of left side. Head circum-ference at birth was 24.5 cm (expected 32 cm) and crown rump length was 23.7 cm (corresponding to 27 weeks). The external genitalia were normal male. Baby was alive for 11 hours. Necrogram was normal except for scattered intracranial calcification. Internal examination showed dilated ventricles and cisterns including posterior fossa cyst in CNS. Cerebellum and vermis were hypoplastic. White caseous material was seen on cerebral ventricular walls all over. Small and scattered white firm areas were seen in cerebral tissue as well. No other abnormaility was seen in thorax, abdomen and umbilical cord. The placenta was remarkably small, however without any abnormality. Chromosomal analysis was normal male (46, XY). TORCH (rubella, cytomegalo virus, toxoplasma and herpes simplex type 2 virus) analysis from cord blood sample was negative for IgM. IgG values were 10-15% less of maternal values indicating trans-placental transfer of maternal IgG antibodies. Brain tissue (from caseous material) subjected to PCR analysis with primer specific for CMV showed negative result. Histopathological examination of brain showed markedly thinned out cortical mantle and ill-formed gyral pattern. All the neuronal elements appeared immature (oligodendro-glial like cells) without any evidence of pyramidal/multipolar neurons with dendritic branching. In addition to defects in neuronal maturation and differentiation, there was evidence of defects in neuronal migration (presence of clusters of small immature round neuronal cells in the deep white matter). The cell density was low and without mitotic activity. The cortical ribbon was infarcted and showed foci of calcification as well as foamy histiocytes (Fig. 1). Many foamy histiocytes were filled with calcific material. The lower part of cortex and white matter was transversed by numerous branching vascular channels with prominent endothelium. Wall of these capillaries were thickened and focally hyzlinized (Fig 2).The vascular proliferation with neovascularization and ischemic changes were focal. Islands of spotty and flaky calcification were seen in white matter, whereas laminar calcification was seen in cortex. Calcification was not seen in the vessels. The leptomeninges were normal, having dilated vascular channels, normal for the age. The leptomeninges were firmly apposed to the pia and developing cortex.
In white matter, many histiocytes were seen. Reactive astrocytes were also seen along the margin of ischemia. In addition, focal globular islands of oligodendroglial like cells were seen. The ependymal lining of lateral ventricle showed attenuation. The subependymal vessels were prominent, engorged and some contained fibrin thrombi. The subependymal zone was gliotic, the reactive gliosis was extending into basal ganglia. The neovascularization noted in cortex was not evident close to ventricle. Extensive calcification and vascular proli-feration was seen in basal ganglia. Tardy neuronal maturation also was seen in basal ganglia. No distinct inflammatory pathology or encephalitis like feature was seen in any area. Geimsa staining was negative for toxoplasmosis. The vascular proliferation and calcifica-tion was also evident in cornea, however, to a lesser extent. The other organs, in particular liver, kidney and lungs did not show such changes. There was no muscle (calf) hypoplasia on histo-pathologic examination. Discussion Fowler et al. in 1972(1) first described hydrencephalic-hydrocephalic syndrome in five female sibs, including in one of a pair of dizygotic twins. Pathological examination revealed remarkable glomeruloid proliferative vasculopathy in central nervous system (CNS) and retina. After rejecting the possibility of a congenital infection, authors proposed an autosomal recessive genetic defect. This report from Australia was subsequently followed by five further publications(2-6) from Australia, Canada, UK, Spain and Belgium, describing similar findings. Here, we describe seventh family of similar syndrome. Our findings, in particular that of CNS, were closely similar to previous reports. The major findings were dilatation of ventricles and disorganized cerebral mantle associated with vascular proliferation and calcification throughout the CNS. Vascular proliferation was not restricted to CNS but was also present in cornea. In contrast to other reports, our case did not show any evidence of arthrogryposis, pterigia, muscular hypoplasia and poly-hydramnios. Instead, it showed severe oligohydramnios and growth restriction. Furthermore, we observed first time in this case clinical findings like depressed pulsatile fontanel, corneal haziness and micro-ophthalmia. This is the third male affected conceptus among 17 cases from seven families. Onset of pathogenesis of our case appears to be after 16 weeks. This is in accordance with Fowler et al., 1972(1). The predominant feature of this syndrome is marked brain immaturity. Though the brain was from 35 weeks fetus, the gyral pattern resembled 15-16 weeks brain. The brain had partially matured as evidenced by the formation of marginal zone (molecular layer) and semblance of laminal cortex. In cortical ribbon many histiocytes filled with spherules of calcification in the cytoplasm were seen suggesting metastatic calcification. Like previous authors(2-6), we regard the calcification as secondary occurring mainly in foci of necrosis. Capillaries were engorged with blood suggesting venous impedance and defective drainage. The vascular proliferation resembled a subacute infarct in evolution in adult, indicating primary vascular pathology in evolution. Although the vessels were prominent, there were few functional lumens due to endothelial hypertrophy. Presence of focal globular islands of oligodendroglial like cells in the white matter indicates defect in migration also. The histologic features of persistence of oligodendroglial like cells in cortex and lack of neuronal maturation suggests that the vascular insult of defective microcirculation had occurred in early second trimester and neurogenesis-glio-genesis remained immature. Absence of calcification in vascular component and pathologic process away from outer cortex-meninges excludes possibility of Sturge-Weber syndrome(7). Absence of distinct inflammatory pathology or encepha-litis like features excludes possibilities of intrauterine infection. This possibility was also excluded by appropriate microbiological studies. The mechanism of ventriculomegaly in this syndrome is not clear. The progressive genesis starting from lateral ventricles indicates that mechanism of ventricular dilatation was not due to obstruction like aqueductal stenosis (also supported by autopsy) but rather due to primary progressive destruction of cerebral cortex. Later, after birth, this was again supported by findings of depressed anterior fontanel (reduced CSF pressure). Our findings indicate that ventricular dilatation was primarily due to failure of CNS development along with parallel destruction of CNS due to vascular pathology. In contrast to other reported cases, our case did not have fetal akinesia sequence (muscle hypoplasia, less fetal movement, arthro-gryposis, pterigia, polyhydramnios, etc.) but had extreme oligohydramnios and growth retardation. This heterogenity could be due to less extensive affection of anterior horn cells. Our serial ultrasound scan indicates that pathogenetic process in our case was maximally operative after 16 weeks and probably late second trimester to early third trimester. This is against the views of other authors(3,4,6) who proposed that patho-genesis starts as early as first trimester. The importance of this proliferative vasculopathy-hydrocephaly case report is its association with extreme oligohydramnios, growth restriction and absence of myopathy, which has not been earlier reported with this syndrome. This case warrants for definition of minimum criteria for the diagnosis and it appears that polyhydramnios and muscle hypoplasia are not essential to label this syndrome. At present it seems a hetero-geneous entity with autosomal recessive inheritance(1,2,4,6) or mitochondrial respi-ratory chain defect(5). Acknowledgement The authors thank Prof. SK Shankar, Head, Department of Neuropathology, National Institute of Mental Health and Neuro-science’s, Bangalore for his expert CNS histopathological evaluation. The authors also thank Prof. S S Agarwal, ex head, Medical Genetics, SGPGIMS, Lucknow for his critical reading of the manuscript and providing some scientific input. Authors are grateful to Dr. Kamal Kishore, Additional Professor, Depart-ment of Microbiology, SGPGIMS and Dr. Anju Rani, Gynecologist, General Hospital, SGPGIMS for their role in the management of the patient. Contributors: AH suspected the conditon, coordinated work-up, carried out investigations and drafted the paper; he will act as the guarantor of the paper. IP was involved in fetal autopsy. LP carried out histopathological examinations. She had also coordinated cross-examination from a reputed neuropathologist (Prof. S.K. Shankar). Funding: None. Competing Interests: None stated.
| ||||
|
References | ||||
|
![]()