|
|
|
Indian Pediatr 2009;46: 532-534 |
 |
Carbonic Anhydrase II Deficiency: A Novel
Mutation |
|
Sheela Nampoothiri and Yair Anikster*
From the Department of Pediatric Genetics, Amrita
Institute of Medical Sciences & Research Center, Aims Ponekkara, PO,
Cochin, Kerala, India;
and *Metabolic Disease Unit, Safra Children Hospital, Sheba Medical
Center, Tel – Hashomer, Israel.
Correspondence to: Dr Sheela Nampoothiri, Consultant,
Department of Pediatric Genetics, Amrita Institute of Medical Sciences &
Research Center, Aims Ponekkara PO, Cochin 682041, Kerala, India. E mail:
[email protected]
Manuscript received: February 21, 2008;
Initial review: March 14, 2008;
Accepted: June 5, 2008.
|
|
Abstract
Carbonic anhydrase II (CA II) deficiency is an
extremely rare autosomal recessive disorder, characterised by a triad of
osteopetrosis, renal tubular acidosis and cerebral calcifications. A
12-year-old boy with classical features of CA II deficiency is reported
who was found to be homozygous for the mutation in CA II gene and
parents were heterozygous for the same mutation .To the best of our
knowledge this is the first case report of mutation proven CA II
deficiency from India.
Key words: Carbonic anhydrase II deficiency, Intracranial
calcifications, Osteopetrosis, Renal tubular acidosis.
|
|
C
arbonic anhydrase II (CA II)
deficiency is a rare autosomal recessive disorder leading to the
development of osteopetrosis, renal tubular acidosis (RTA) and
intracranial calcification.
Case Report
A 12-year-old boy who was diagnosed as having
osteopetrosis at 8 months of age was brought for evaluation. The child was
receiving low dose corticosteroid therapy from 8 months of age. At the age
of 8 ½ years he was found to have short stature. Steroids were stopped and
detailed evaluation revealed renal tubular acidosis. Other hormonal causes
of short stature were ruled out. He was started on oral potassium citrate
and was periodically evaluated.
Presently the parents had brought him to the Genetics
department as they were planning another baby. The couple had a younger
girl child who had increased bone density and died at the age of 5 years
following drowning. On evaluation at 12 year, he was studying in 5th
standard and has scholastic backwardness. His weight was 19.5 kg, height
116.5 cm and head circumference 49 cm (all were below third centile). He
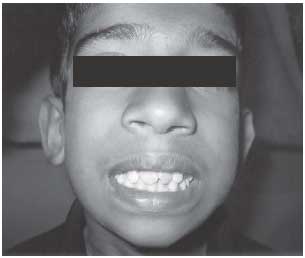
did not have any dysmorphic features apart from overcrowded teeth (Fig.1).
 |
|
Fig. 1 Proband with over crowded
teeth. |
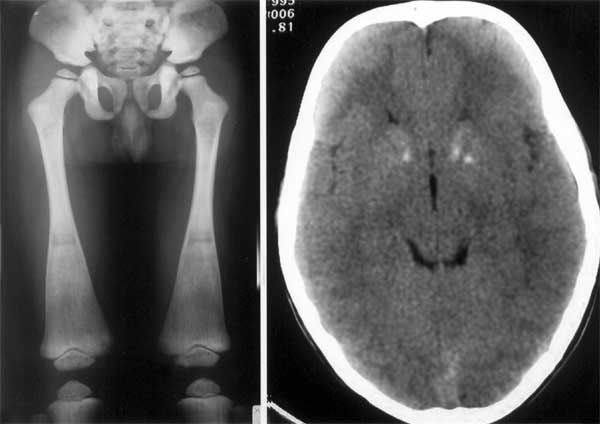
Skeletal survey showed hyperdensity of bones compatible
with osteopetrosis with flared metaphyses of femurs (Fig. 2a).
Ophthalmologic evaluation showed bilateral posterior subcapsular
cataracts, secondary to prolonged steroid therapy. Fundus evaluation and
audiogram were normal. His laboratory results showed metabolic acidosis
(pH 7.32, bicarbonate 16.3 mmol/L, sodium 138 mmol/L, potassium 3.9 mmol/L,
chloride 110 mmol/L, urine pH 6.3, anion gap 11.7, creatinine 0.7 mg/dL).
All features were consistent with classical renal tubular acidosis.
Ammonium chloride loading test failed to reduce the urine pH to less than
6 suggesting distal RTA. Hematological work up showed hemoglobin of 12.9
g/dL, WBC 8650/cc, platelet count of 2,79000/cc with a normocytic normo-chromic
picture in the peripheral smear.
 |
|
Fig. 2a Osteopetrosis of the femur with flared
metaphysis at the lower end. |
Fig. 2b CT scan of brain showing bilateral basal
ganglia calcifications.
|
A combination of osteopetrosis with distal renal
tubular acidosis raised the possibility of carbonic anhydrase II
deficiency. Acid phosphatase was found to be elevated 7 U/L (normal up to
5.1). Abdominal ultrasound scan was normal whereas brain CT scan showed
calcification of both caudate nuclei and left putamen (Fig.2b).
With these evaluations the diagnosis of Carbonic anhydrase II deficiency
was confirmed. DNA was extracted from peripheral blood samples of the
proband and parents. Genomic DNA of the patient was then PCR amplified for
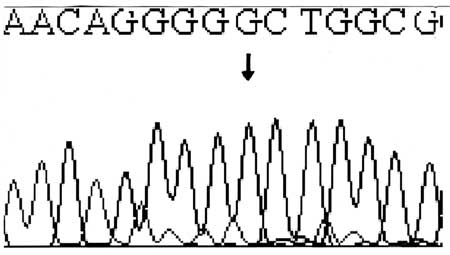
each of seven exons of the CA2 gene and analysed by direct nucleotide
sequencing. A novel T85C substitution that causes the S29P (serine to
proline) amino acid change was identified (Fig.3). This
missense mutation in exon 2 had destroyed a Bsr1 restriction site in the
mutant amplicon. Using this restriction enzyme, the parents were checked
for this novel mutation and they were found to be heterozygous. Treatment
is being continued with potassium citrate and the biochemical parameters
and growth is being monitored periodically. Mother is now 8 weeks pregnant
and we are planning to offer prenatal diagnosis for this couple.
 |
|
Fig. 3 Reverse sequencing showing T>C
substitution at position 85 in the exon 2 in genomic DNA. |
Discussion
The clinical course of CA II deficiency is benign and
this condition is compatible with long-term survival and hematological
abnormalities associated with lethal form of osteopetrosis are absent in
CA II deficiency(1). This condition is called as marble brain disease as
these patients present with osteopetrosis and cerebral calcifications(2).
Carbonic anhydrase gene has been mapped on chromosome 8q 22. Carbonic
anhydrase is a zinc metalloenzyme and catalyses reversible hydration of CO 2.
CA II has a wide tissue distribution and is found in bone, kidney
(proximal tubule and collecting duct), erythrocytes, glial cells and
osteoclasts(3). The facial features of CAII deficient patients include
broad forehead, small mouth, delayed dentition and dental malocclusion(1).
These patients have failure to thrive and growth retardation as universal
features. Around 50% of the patients have mild mental retardation.
CAII is involved in the proximal renal tubular
bicarbonate reabsorption and distal renal tubular acidification. Hence the
patients with CA II deficiency have characteristics of both proximal and
distal renal tubular acidosis(4). There is increased tendency for
nephrocalcinosis in patients who have predominant distal RTA. Deficiency
of CA II impairs the production of H +
ions by osteoclasts thereby blocking the resorption of bones and leads to
development of osteopetrosis(3). As the balance between osteoclast and
osteoblast activity is disrupted the excessive accumulation of brittle
bone leads to osteopetrosis and increased risk for fractures. All patients
have increased density of axial skeleton, long bones and skull along with
widening of metaphysis of long bones. These patients have normal levels of
calcium and phosphorus, mildly elevated alkaline phosphatase and acid
phosphatase.
Intracranial calcifications in CAII deficiency are not
usually present at birth and they usually develop by 2-5 years of age.
Calcifications are seen in basal ganglia and also in cortex. Basal ganglia
calcifications primarily appear in putamen and caudate nucleus.
Calcification of cortex initially involves frontal lobes(5). The reason
for intracranial calcification is still unclear. Cranial nerve entrapments
leading to blindness and deafness are rare in CA II deficiency(1).
Restrictive lung disease has also been observed in few patients. Hence
periodic pulmonary function tests can detect these complications early(6).
There is no role for corticosteroids in CAII deficiency
due to relative lack of hematological complication. Allogenic bone marrow
transplan-tation has a role in the attenuation of osteopetrosis and
cerebral calcification but has no effect on renal tubular acidosis and
mental retardation(1,3,7).
Early institution of alkali supplementation is the
sheet anchor in the management of renal tubular acidosis as RTA is the
most likely cause for growth retardation.The patients who were diagnosed
and treated with sodium bicarbonate before the age of 4 years have
attained normal adult height(1). CA II deficiency should be considered in
children presen-ting with osteopetrosis without anemia and
thrombo-cytopenia so that long-term steroid therapy can be avoided.
Prenatal diagnosis could be offered either from cultured amniocytes or
from chorionic villus biopsy by direct sequencing provided the mutation in
the proband is identified beforehand(8, 9).
Contributors: SN diagnosed the condition in
the index case, evaluated the family tree and drafted the article. She
will act as the guarantor of the manuscript. YA has done the molecular
genetic analysis for the family for confirmation of the diagnosis and has
contributed to the drafting.
Funding: None.
Competing interests: None stated.
References
1. Awad M, Al Ashwal AA, Sakati N, Al-Abbad AA, Bin
Abbas BS. Long-term follow up of carbonic anhydrase II deficiency
syndrome. Saudi Med J 2002; 23: 25-29.
2. Sly WS, Hewett-Emmett D, Whyte MP, Yu Y-SL, Tashian
RE. Carbonic anhydrase II deficiency identified as the Primary defect in
the autosomal recessive syndrome of osteoporosis with renal tubular
acidosis and cerebral calcification. Proc Nat Acad Sci USA 1983; 80:
2752-2756.
3. Cotter M, Connell T, Colhoun E, Smith OP, McMahon C.
Carbonic anhydrase II deficiency: a rare autosomal recessive disorder of
osteopetrosis, renal tubular acidosis, and cerebral calcification. J
Pediatr Hematol Oncol 2005; 27: 115-117.
4. Bolt RJ, Wennink JM, Verbeke JI, Shah GN, Sly WS,
Bökenkamp A. Carbonic anhydrase type II deficiency. Am J Kidney Dis 2005;
46: 71-73.
5. Cumming WA, Ohlsson A. Intracranial calcifi-cation
in children with osteopetrosis caused by carbonic anhydrase II deficiency.
Radiology 1985; 157: 325-327.
6. Lotan D, Eisenkraft A, Jacobsson JM, Bar-Yosef O,
Kleta R, Anikster Y, et al. Clinical and molecular findings in a
family with the carbonic anhydrase II deficiency syndrome. Pediatr Nephrol
2006; 21: 423-426.
7. McMahon C, Will A, Hu P, Shah GN, Sly WS, Smith OP.
Bone marrow transplantation corrects osteopetrosis in the carbonic
anhydrase II deficiency syndrome. Blood 2001; 97: 1947-1950.
8. Shah GN, Bonapace G, Hu PY, Strisciuglio P, Sly WS.
Carbonic anhydrase II deficiency syndrome (osteopetrosis with renal
tubular acidosis and brain calcifications): Novel mutations in CA2
identified by direct sequencing expand the opportunity for
genotype-phenotype correlation. Hum Mutat 2004; 24: 272.
9. Strisciuglio P, Hu PY, Lim EJ, Ciccolella J, Sly WS. Clinical and
molecular heterogeneity in carbonic anhydrase II deficiency and prenatal
diagnosis in an Italian family. J Pediatr 1998 ; 132: 717-720.
|
|
|
 |
|
