|
|
|
Indian Pediatr 2011;48: 559-561 |
 |
Cartilage-hair Hypoplasia Caused by Novel
Compound Heterozygous RMRP Mutations |
|
Kerstin Reicherter, Amithkumar Iynapillai Veeramani*
and Sujatha Jagadeesh*
From Center for Pediatrics and Adolescent Medicine and
Faculty of Biology, University of Freiburg, D-79106 Freiburg, Germany;
and*Department of Clinical Genetics, Fetal Care Research Foundation,
Chennai, India.
Correspondence to: Dr Sujatha Jagadeesh, Consultant and
Head, Department of Clinical Genetics,
Fetal Care Research Foundation, Chennai, India.
Email: [email protected]
Received: June 23, 2009;
Review Completed: January 11, 2010;
Accepted: March 22, 2010.
|
Cartilage-hair hypoplasia is a rare, autosomal recessive skeletal
dysplasia, caused by mutations in the RMRP gene. The skeletal
abnormalities include irregular metaphyses and cone shaped epiphyses of
the hands. Molecular diagnosis confirmed two novel RMRP mutations
in a compound heterozygous state in two siblings with this condition.
Key words: India, Metaphyseal chondrodysplasia, RMRP mutation,
Skeletal dysplasia.
|
|
Cartilage-hair
hypoplasia (CHH) or metaphyseal chondrodysplasia McKusick type (OMIM
#250250) is an autosomal recessive disorder [1]. It is a type of skeletal
dysplasia resulting in short-limbed dwarfism with shortening and sometimes
bowing of the tubular bones and brachydactyly, with radio-graphs showing
disorganized metaphyses as well as cone shaped epiphyses of the fingers
[2]. The RMRP gene was identified as the disease causing gene in
CHH [3,4]. We report two patients with CHH caused by the two novel RMRP
mutations c.94_96dupAGT and c.99C>T.
Case Report
Two children born to a third degree consanguineously
married South Indian couple were referred for evaluation of short stature.
The elder sibling was a 15-year-old girl with short fingers at birth.
Later a delay in all motor milestones and a gradual loss of scalp hair
were noticed. She was attending school and was an average performer. When
clinically evaluated at the age of 15-yr, the weight was 21 kg (<5 th
percentile) and height was 108 cm (<3rd percentile). The upper and lower
segment ratios were infantile with upper segment measuring 57 cm while the
lower segment measured 51 cm; the arm span (118 cm) was greater than her
height. Other skeletal abnormalities included pectus carinatum,
exaggerated lumbar lordosis, genu valgum and pes planus. The
extension of elbows was restricted. Brachydactyly with broad finger tips
and a single crease in the thumbs were identified. There was no axillary
hair and she had not yet attained menarche. There was no history
suggestive of immunodeficiency or malabsorption. Radiographs revealed cone
shaped epiphyses of the hands, metaphyseal irregularities of femoral heads
and phalanges, and brachydactyly of the phalanges.
The younger sibling was a 9-year-old boy whose height
(99 cm) and weight (18 kg) were below the 5 th
percentile. His phenotype was similar to that of his sister, except for
less pronounced scalp hair loss. Radiographs revealed similar changes;
metaphyseal dysplasia and platyspondyly of the thoracolumbar vertebra was
also seen. The patient’s radiographs were submitted to the European
Skeletal Dysplasia Network (ESDN) clinical-radiographic review panel. As
the features were suggestive of CHH, analysis of the RMRP gene was
proposed.
Following extraction of DNA from blood of the sibs and
their parents, the RMRP gene was amplified by polymerase chain
reaction (PCR) [1]. The complete transcript of the RMRP gene as
well as 200 base pairs of the promoter region was sequenced. Thereby the
novel RMRP mutations c.94_96dupAGT and c.99C>T (Fig. 1)
as well as the previously described polymorphisms g. 58T>C; g.-48C>A and
g.+7T>C (all homozygous), were identified in both patients. Both siblings
were compound heterozygous for the mutations. The correct segregation of
the mutations was proven by subcloning and sequencing of the PCR products.
Analysis of the parents confirmed their carrier status. The father had a
c.99C>T mutation whereas the mother had the duplication (c.94_96dupAGT).
The father was homozygous for the polymorphisms and the mother was
heterozygous.
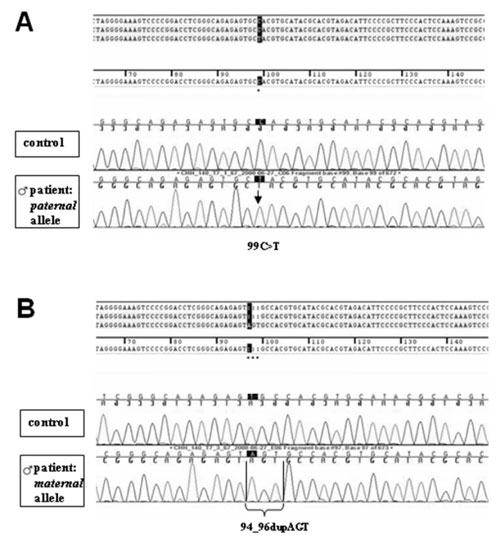 |
|
Fig 1. Sequencing of the RMRP gene after
subcloning; results of the boy. (a) The paternal allele carries a
c.99C>T nucleotide substitution within the RMRP gene. (b) The
maternal allele carriers a duplication of 3 nucleotides
(c.94_96dupAGT) within the RMRP gene. Both patients were compound
heterozygous for the novel RMRP mutations c.99C>T and c.94_96dupAGT. |
Discussion
CHH is a rare disease with known carrier frequencies of
1:19 among the Amish and 1:76 among the Finnish population [3]. The
phenotype of CHH is variable. Laxity of ligaments, incomplete extension at
the elbows, cone shaped epiphyses of the phalanges [1], rounded distal
epiphyses, prominent sternum, chest deformity [5], mild scoliosis,
increased lumbar lordosis and bowing of the lower limbs are described. The
affected siblings reported in this study, had most of the clinico-radiographic
findings suggestive of CHH.
The RMRP gene encodes the untranslated
RNA component of the RNase MRP complex. This ribonucleoprotein complex
cleaves RNAs and is thereby involved in the replication of mitochondrial
DNA [6], processing of the 5.8 S ribosomal RNA [7] and control of cell
cycle progression at the end of mitosis [8]. Mutations in the RMRP
gene lead to a wide spectrum of recessive skeletal dysplasias with
different degrees of short stature. The molecular analysis of the RMRP
gene in the affected siblings identified two novel mutations,
c.94_96dupAGT and c.99C>T. We conclude that these were pathogenic as they
were inherited from unaffected parents, both carrying one of the
mutations. In addition, the mutations were not identified in 100
controls [9]. The functional importance could be explained by the
localization of both mutations in an evolutionary conserved part of the
transcript [9]. They might alter a very important stem-loop structure
[10].
After having confirmed the diagnosis by molecular
analysis, the prospective treatment was discussed. Unlike most CHH
patients, these sibs did not have a history of recurring infections or
other signs of immunodeficiency. As the clinical outcome of CHH patients
can only be poorly predicted, they should be carefully followed up because
of the possibility of serious infections and malignancies.
Acknowledgments: We acknowledge the support
of Dr Andreas Zankl, Dr Sheila Unger and the ESDN clinical-radiographic
review panel (www.esdn.org). We thank Dr Pia Hermanns, who was involved in
the RMRP mutation analysis and the interpretation of the data. We
thank Dr Suresh S of the Fetal Care Research Foundation for his
encouragement.
Contributors: KR: did the molecular analysis, was
involved in the interpretation of the data, drafted the manuscript
together with AIV and revised the manuscript; AIV: drafted the manuscript
initially; SJ: managed the case and will act as the guarantor. The final
manuscript was approved by all authors.
Funding: None.
Competing interests: None stated.
References
1. Bonafé L, Schmitt K, Eich G, Giedion A,
Superti-Furga A. RMRP gene sequence analysis confirms a cartilage-hair
hypoplasia variant with only skeletal manifestations and reveals a high
density of single-nucleotide polymorphisms. Clin Genet. 2002;61:146-51.
2. Polmar SH, Pierce GF. Cartilage hair hypoplasia:
immunological aspects and their clinical implications. Clin Immunol
Immunopathol. 1986;40:87-93.
3. Ridanpää M, Sistonen P, Rockas S, Rimoin DL, Mäkitie
O, Kaitila I. Worldwide mutation spectrum in cartilage-hair hypoplasia:
ancient founder origin of the major 70A->G mutation of the untranslated
RMRP. Eur J Hum Genet. 2002;10:439-47.
4. Ridanpää, M, van Eenennaam H, Pelin K, Chadwick R,
Johnson C, Yuan B, et al. Mutations in the RNA component of RNase
MRP cause a pleiotropic human disease, cartilage-hair hypoplasia. Cell.
2001;104:195-203.
5. McKusick VA, Eldridge R, Hostetler JA, Egeland JA,
Ruangwit U. Dwarfism in the Amish. II Cartilage-hair hypoplasia. Bull
Johns Hopkins Hosp. 1965;116:285-326.
6. Chang DD, Clayton DA. Mouse RNAase MRP RNA is
encoded by a nuclear gene and contains a decamer sequence complementary to
a conserved region of mitochondrial RNA substrate. Cell. 1989;56:131-9.
7. Schmitt ME, Clayton DA. Nuclear RNase MRP is
required for correct processing of pre-5.8S rRNA in Saccharomyces
cerevisiae. Mol Cell Biol. 1993;13:7935-41.
8. Gill T, Cai T, Aulds J, Wierzbicki S, Schmitt ME.
RNase MRP cleaves the CLB2 mRNA to promote cell cycle progression: novel
method of mRNA degradation. Mol Cell Biol. 2004;24:945-53.
9. Bonafé L, Dermitzakis ET, Unger S, Greenberg CR,
Campos-Xavier BA, Zankl A, et al. Evolutionary comparison provides
evidence for pathogenicity of RMRP mutations. PLoS Genet. 2005;1:444-54.
10. Welting TJ, van Venrooij WJ, Pruijn GJ. Mutual
interactions between subunits of the human RNase MRP ribonucleoprotein
complex. Nucleic Acids Res. 2004;32:2138-46.
|
|
|
 |
|
