Jayashree Muralidharan*
Nandita Kakkar
B.D. Radotra
From the Departments of Pediatrics * and Histopathology, Postgraduate Institute
of Medical Education and Research, Chandigarh 160012, India.
Reprint requests: Dr. B.D. Radotra, Associate Profes- sor, Department of Histopathology, PGIMER Chandigarh 160012, India.
E-mail:
[email protected]
A female child born at 34 wks of gestation by spontaneous vaginal delivery in the Post Graduate Institute of Medical Education and Research (PGlMER) on 4.7.1998 at 9.04 AM with a birth weight of 3.1 kg, had problems of
prematurity, respiratory distress and congenital malformations as described below:
The baby was fifth in birth order and was born to a 28 years old 5th gravida mother who was married for last 13 years. The first pregnancy, 12 years back, was unsupervised and a female child was born at term by assisted breech and is presently doing well. The second pregnancy 10 years back was again unsupervised and a male baby born at term by assisted breech, had problems of birth asphyxia and
died at 5 min of birth. The baby was also noticed to have underdeveloped genitalia. The third was born preterm at 37 weeks by assisted breech, had laboured
breathing, microphthalmia and CTEV and died 5 min after birth. The fourth pregnancy was supervised and mother went into preterm labour at 33 weeks, delivering a male child weighing 2.7 kg, with problems of birth asphyxia and respiratory distress syndrome. No external congenital malformations were noticed. The autopsy
of this
baby
had revealed features of HIE stage
III, with meconium aspiration syndrome (MAS) with bilateral polycystic kidney disease and pericardial cyst.
The present pregnancy was booked and supervised from 8 weeks onwards at PGIMER. No medical problems were noted during pregnancy. Antenatal ultrasound (USG) at 24 weeks had revealed bilateral polycystic kidneys with oligohydramnios. Bladder was not visualized during USG examination.
The baby was born preterm at 34 wks and had birth asphyxia with Apgar score of 2 and at 1 min and 5 min, respectively. Resuscitation with bag and mask for 15 sec was followed by intubation
and manual ventilation. She required cardiac compressions for 10 sec and ETT adrenaline during resuscitation. The placental weight was 1 kg. The respiratory distress which started soon after birth continued till the demise. Manual ventilation with 100% oxygen was continued. The chest expansion was noted to be poor on pressures approximately 40 cm of HP and the Sa02 was between 60-70%. Examination revealed a gestational age of 34 weeks with occipito frontal circumference of 32.3 cm. The left ear was malformed but there were no other obvious
dysmorphic features. Abdominal examination revealed bilateral renal masses,
though no further delineation or description of this mass is available as per the case records. The cardio- vascular system and chest were within normal limits. Postnatal abdominal ultrasound showed bilateral grossly enlarged kidneys with in- creased medullary echogenicity. No definite cysts were seen. The urinary bladder was empty at the time of examination. The liver was reportedly normal and lungs were isoechoeic with the diaphragm. The course in the hospital was mainly supportive with IV fluids and manual ventilation. The baby succumbed to her problems on the same day and died after a hospital stay of seven hours.
Clinical Discussion by Dr. Jayashree
To analyze, we have a case with history of previous sibling deaths, bilateral enlarged kidneys, clinical and radiographic evidence of pulmonary hypoplasia supported by antenatal and postnatal ultrasonogram
suggestive of bilateral renal disease. In all probability we are
dealing with a congenital and heritable renal disorder. Since the
disease has affected both the sexes and in the absence of parental
screening, the mode of inheritance can be either autosomal recessive or dominant. Renomegaly was the most important clinical clue in this child. The causes of renal masses in newborn (Table /) reveal that hydronephrosis
is commonest followed by multicystic dysplasia and polycystic kidney disease(l). Hydronephrosis, polycystic kidneys and other cystic diseases are usually bilateral whereas multicystic dysplasia, tumors and renal vein thrombosis are mostly unilateral. Bilaterality is rare in the latter group. Bilateral hydronephrosis with an empty bladder suggests a pelviuretic obstruction. Pelviuretic junction (PVJ) obstructions are by and large unilateral with less than 10% presenting as bilateral masses(2,3). Familial variety of hydronephrosis is a condition postulated to be due to PUJ obstruction or to a faulty muscle development. Ultrasound showing dilated calyceal system is diagnostic for hydronephrosis. Multicystic dysplasias
are second commonest cause for renal enlargement and majority are unilateral. It
occurs sporadically and recurrences are rare. The kidney architecture is distorted by large lobulated cysts which represent the failure of communication between the secretory and excretory portion of the nephron. Associated ureteral anomalies are usually seen. USG reveals large globular cysts and is diagnosic of this entity. In absence of the typical ultrasound findings as described above both hydronephrosis and multicystic dysplasias
are ruled out in this index case.
Table 1
Differential Diagnosis of Enlarged Kidneys in Neonates (n = 191)
|
Lesion |
Number |
% |
| Hydronephrosis |
74 |
39 |
| Multicystic dysplasia |
59 |
31 |
| Polycystic Kidneys |
16 |
8 |
| Ectopia |
12 |
6 |
| Tumor |
10 |
5 |
| Renal vein thrombosis |
5 |
3 |
| Horse shoe kidney |
4 |
2 |
| others |
11 |
6 |
"The more complicated an organ in its development the more subject it is to
mal-development and in this respect the kidney outranks most other organs" (Edith L. Potter).
Polycystic kidney disease (PKD) accounts for 8% of renal masses in newborn. It is con- ventionally divided into autosomal recessive polycystic kidney disease (ARPKD)
and autosomal dominant polycystic kidney disease (ADPKD). ARPKD is seen in 1: 1 0,000 and has a varied spectrum of presentation ranging from perinatal, neonatal, infantile to juvenile
forms(4). Renal disease predominates over liver involvement in patients where disease develops at birth or during the neonatal period. It involves an embryonic defect in which there is a diffuse dilation of the collecting tubules. Presence of congenital hepatic fibrosis is a must for the diagnosis. ADPKD is common in adults with an incidence of I: 1000, but is being increasingly recognized in children over
2-3 decades. In one longitudinal study the incidence of ADPKD in children less than one I year was found to be 12%(5). It involves cystic dilation of any part of nephron, including
glomerulus. Clinical spectrum in perinatal and neonatal presentation can be similar in both and differentiation on clinical grounds can be extremely difficult. The only investigative tool at our disposal is the ultrasound which shows enlarged kidneys, enhanced medullary echogenicity and no definite cysts. These sonographic findings can very well go with the diagnosis of PKD. However, sonogram can be similar in both ARPKD and ADPKD during neonatal period with some subtle differences as seen in Table II. The presence of macrocysts
(>2 cm) and a fairly well delineated renal margin due to cysts in the peripheral most cortex are some of the sonographic features more often seen in ADPKD than ARPKD(4,6).
Table II
Sonographic Features of Recessive Polycystic Kidney Disease
| Age group
|
Sonographic Features |
| Neonate |
Massive Kidney Enlargement |
| |
Increased echogenicity of
entire parenchyma |
| |
loss of corticomedullary
differentiation |
| |
loss of central echo complex |
| |
small macrocysts less than 2 cm
in diameter |
| Children |
Massive kidney enlargement |
| |
Increased echogenicity in
medulla |
| |
Macrocysts less than 2 cm. |
| |
Hepatic cysts |
| |
Pancreatic Cysts |
| |
Splenomegaly secondary to
portal hypertension. |
From the clinical and sonographic findings very little can be done to differentiate between the two forms ofPKD in its perinatal-neonatal presentation. Few other conditions can mimic radiological appearance of polycystic kidney disease (Table III). PKD associated with Meckel's, Jeune's and Ivemark syndrome can be differentiated by associated phenotypic features which are part of the syndrome com- plexes. PKD associated with tuberous sclero- sis is unique in that the renal manifestation may antedate the neurocutaneous
features. There are case reports of renal disease beginning . in utero itself(7). In absence of other
clinical pointers and parental screening, entertaining this diagnosis is not possible. The familial hypoplastic glomerocystic kidney and congenital hypemephronic nephromegaly are mostly histopathological diagnosis.
Table III
Differential Diagnosis of Cystic Kidney Disease in Infancy
| 1. |
Autosomal recessive polycystic
kidney disease. |
| 2. |
Autosomal dominant polycystic
kidney disease. |
| 3. |
Bilateral multicystic kidneys. |
| 4. |
Polycystic kidneys associated
with tuberous sclerosis |
| 5. |
Meckel's syndrome |
| 6. |
Ivemark syndrome |
| 7. |
Jeune syndrome (asphyxiating
thoracic dystrophy) |
| 8. |
Familial hypoplastic
glomerulocystic kidney. |
| 9. |
Congenital hypernephronic
nephromegaly with tubular dysgensis |
If the case is PKD, then what extrarenal manifestations does one expect? Congenital hepatic fibrosis with bile ductular dysgenesis is a diagnostic feature of ARPKD and will be found in 100% cases. Hepatic fibrosis has been reported in 10% of ADPKD. Pancreatic cysts, berry aneurysms, endocardial
fibroelastosis and pyloric stenosis are some of the other extrarenal manifestations and when seen are more often a feature of ADPKD than ARPKD. The ratio of baby weight to placental weight was 1:3 as against a normal of 1 :5. Hyperplacentosis which is commonly described with immune and nonimmune hydrops has not been described with PKD. In an autopsy review of 7 cases with PKD, large placenta was reported in one case(8).
The lungs were the other major organs involved in this baby. Severe birth asphyxia, poor chest expansion with manual ventilation, chest X-ray showing bilateral white out and USG showing isoechoeic lungs, suggest the presence of pulmonary hypoplasia.
Occurrence of lung hypoplasia in bilateral severe renal disease is a part of the oligohydramnios sequence. Inhibition of lung growth due to lack of inhaled fluid and uterine compression leading to poor chest expansion are the main reasons for development of hypoplasia. The other findings one expects to find are some- times lung cysts, pneumothorax and interstitial emphysema. the latter can be a complication of resuscitative procedure.
The terminal event has been a respiratory failure, hypoxia and death. The final clinical diagnosis is:
(a)
Autosomal recessive polycystic kidney disease, pulmonary hypoplasia, birth asphyxia and prematurity.
(b)
Autosomal dominant polycystic kidney disease.
Comments Before Pathology Presentation
Prof. V. Sakhuja (Chairmau): Thank you Dr. Jayashree. May I invite comments from the treating unit first.
Dr. Akhil: I have been treating this patient in intensive care unit. She was a preterm neonate who sustained birth asphyxia and severe respiratory distress requiring intermittent positive pressure respiration. There was overtly distended abdomen with bilateral renal masses and genital edema. Due to the short stay we could not investigate this baby as we would have liked to do. However, an ultrasound was done, which revealed involvement of both the kidneys. The capsule of the kidneys was intact. The pelvi-calyceal
system was poorly delineated and the corticomedullary differentiation was less
than optimal. The entire kidneys
were hyperechoiec and bladder was not visualized. The liver was also hyperechoiec, but there were no hepatic cysts. The lungs were isoechoiec
with the diaphragm, which was consistent with bilateral massive atelectasis, sug- gesting pulmonary hypoplasia. The presence of poorly defined, but uniform echo- texture of the kidneys was more consistent with the diagnosis of ARPKD rather than ADPKD. In this case small linearly arranged cysts are likely to be present. In ADPKD, the cysts are usually larger and variable in size, and ultrasound shows variable echotexture. Tuberous sclerosis has been mentioned as one of the causes of cystic disease of kidney, but there are no re- ports of tuberous sclerosis with severe perinatal form leading to pulmonary hypoplasia. Hyperechoic liver in this case indicates some degree of fibrosis. Due to prematurity, there may be hyaline membrane disease. However, oligo-hydramnios and severe renal disease in this patient is likely to be associated with pulmonary hypoplasia. The vulval edema in this case is due to inferior vena cava obstruction be- cause of bilateral renal masses.
Prof. V. Sakhuja: The case is now open for further comments.
Dr. V. Jha: I fully agree with Dr. Jayashree's discussion. But I would only consider ARPKD in this case and not ADPKD using a bit of deductive logic based on information not available in the clinical protocol. There is a history of. autopsy proven PKD in the sib. Therefore, it is not an isolated mutation, but it has to be an inherited disease. I believe that the mother's ultrasound was done but possibly did not show any form of PKD. If it was ADPKD then either mother or father should have PKD. However, one may say that the father of the child has not been screened and it could still be ADPKD.
Prof. V. Sakhuja: I may add more information here that neither mother nor father of this child have been screened by ultrasound. Had they been screened this would have been very useful information. I would also like to point out that since both patients are less than
30
years of age, it is possible that one would have missed the cysts in the parents and therefore in this situation, it is the grandparents who require screening.
Dr. Vanita Jain: The ultrasonographic findings of bilateral large kidneys without any cysts and repeatedly finding of reduced amniotic
fluid right from second trimester onwards point to the diagnosis of ARPKD. In ADPKD, the liquor volume is not so much reduced and usually cysts are seen.
Dr. S.K. Singh: The ARPKD has perinatal, infantile, neonatal and juvenile varieties. Al- though the ultrasound findings favor ARPKD, the incidence is against the diagnosis. In ARPKD, only one out of four children are likely to be affected, whereas in this case all the children have been affected. I think this may be the reason why Dr. Jayashree kept the possibility of ADPKD.
Dr. Jayashree: No, I do not think so. I think inspite of the fact that in ARPKD there is' a 25% incidence of manifestation, the chances for the successive babies getting affected are still there. If one or two babies are involved, the risk for the successive babies increases and therefore, the incidence does not help us in differentiating ADPKD from ARPKD. What is ideally needed in such a situation is parental
screening.
Dr. R.K. Marwaha: I would like to reiterate what Dr. Jayashree has said. In the previous autopsy of the sibling, we have not been shown any hepatic changes such as hepatic fibrosis or biliary dysgenesis. These changes are always shown in the recessive form. If one were to consider that, then ADPKD becomes much likely possibility. The 25% risk of involvement that has been mentioned by Dr. Singh is just like tossing a coin which means that if you get head once, you are not
necessarily going to get tail next time.
Dr. Akhil: I would like to add here that the final autopsy report in the previous baby had revealed presence of hepatic fibrosis.
Prof. V. Sakhuja: May I now request Dr. N. Kakkar to present the pathology findings.
Pathology Protocol by Dr. N. Kakkar (PM No. 16165): A complete autopsy was per- formed on this day 1 female child.
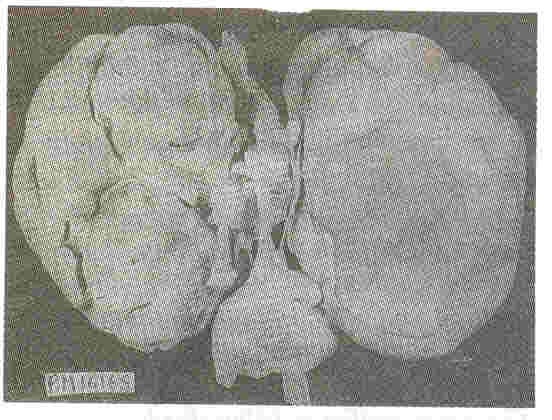
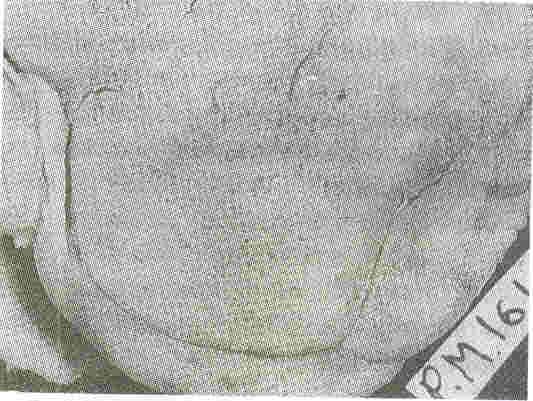
Kidneys: Both kidneys were symmetrically enlarged. The fetallobulations were seen (Fig. 1). The surface was smooth with myriads of pinhead sized cysts recognizable just below the capsular surface (Fig. 2). On cut section the renal substance consisted entirely of radially oriented, elongated and fusiform cysts in the cortex whereas they were more rounded in the medulla (Fig. 3).
 |
 |
| Fig. 1. Symmetrically enlarged
right and left1 kidneys with preserved foetal lobulations. |
Fig.2. External surface of the
kidney at a closer look shows myriads of pinhead sized
cysts |
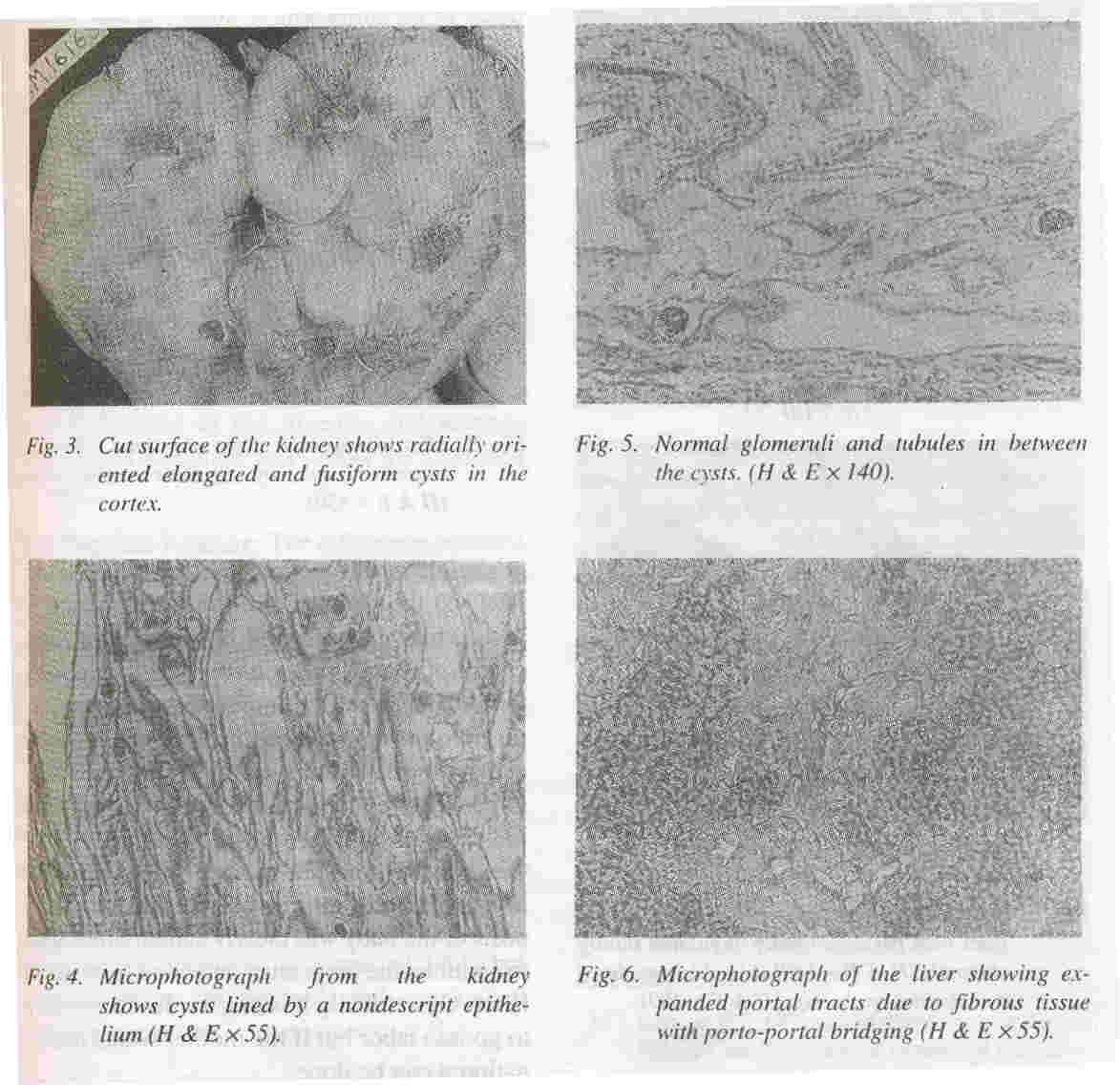 |
Microscopically these radially oriented cysts were lined by low cuboidal nondescript epithelium (Fig. 4). In between the cysts normal tubules and glomeruli were seen (Fig. 5). The glomeruli appeared sparse due to the marked degree of renal enlargement although they are present in normal numbers.
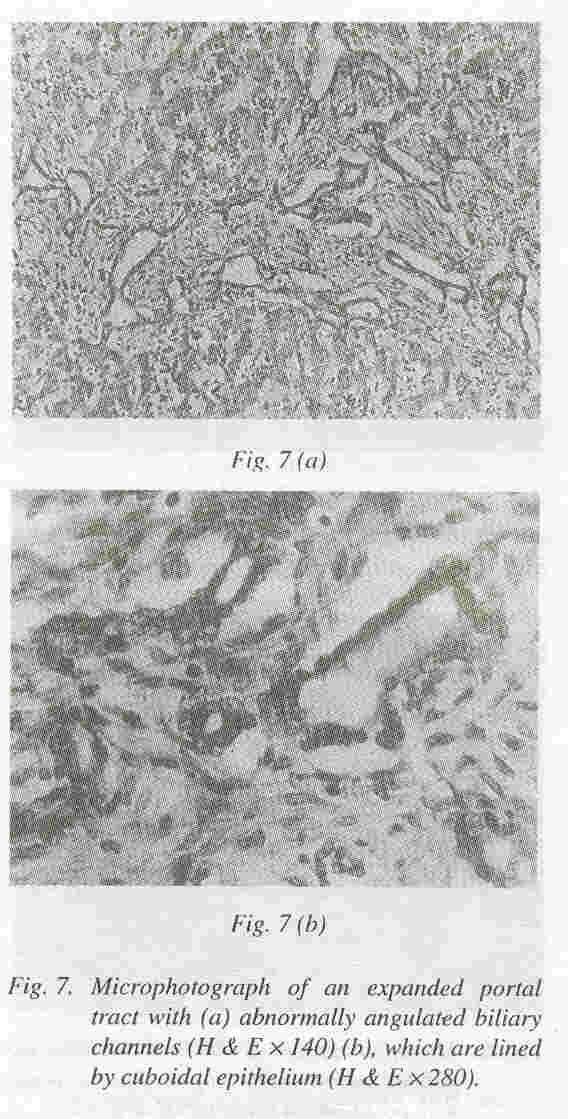
Liver: The liver was enlarged and weighed 130 grams. It was firm in consistency. Microscopially, the portal tract were expanded and showed portal bridging (Fig. 6). The expansion was due to fibrosis and increased number of abnormally angulated biliary channels (Fig. 7a). They exhibited branching and were typically located along the margins of the portal tracts often running parallel to the border. This simulated the embryonic ductal plates suggesting a developmental origin through malformation.
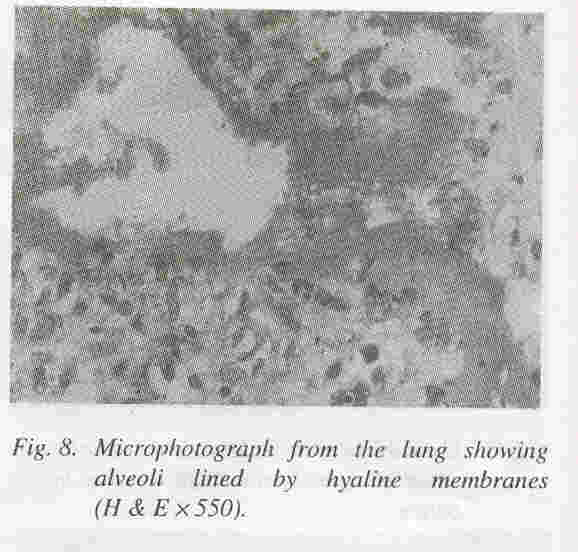
Lungs: The lungs weighed 25 grams and were hypoplastic. Microscopically, there was hyaline membrane disease (Fig. 8). Other organs did not show any significant pathology. The final Autopsy Diagnosis was
*
Autosomal recessive polycystic kidney disease
*
Congenital hepatic fibrosis (ductal plate malformation)
*
Pulmonary hypoplasia and hyaline mem-
.
brane disease.
Grossly and microscopically this was a classical case of recessive polycystic kidney disease with ductal plate malformation. Dominant
polycystic kidney disease can occur in the newborn but bilateral symmetrical
involvement is not seen. The cysts in the dominant form are of variable size, dispersed randomly throughout the cortex and medulla.
RPKD and congenital hepatic fibrosis (CHF) are inseparably intertwined entities. They constitute a single disease process with an extremely wide spectrum of expression. This varies from predominantly renal lesions
with clinically trivial hepatic lessions on one hand to marked portal fibrosis with portal hypertension but only inconspicous asymtomatic
renal cysts on the other hand. Between these two extremes are patients with
variable mixtures and severity of both hepatic and renal problems.
The basic lesion of congenital hepatic fibrosis corresponds to ductal plate malformations of interlobular bile ducts resulting from faulty. development, i.e., disturbance in epitheliomesenchymal inductive interactions.
The immature bile ducts are subject to a progressive destructive cholangiopathy resulting in a pattern of more or less advanced fetal type of biliary fibrosis. The
destructive cholangiopathy may vary in progress and duration in different patients. All cases of ARPKD have CHF but not vice versa.
Sixty per cent case of CHF have ARPKD, however, case reports in literature do show CHF associated with ADPKD, nephronothisis, medullary cystic disease, renal dysplasia and
rarely a normal kidney. Since it is an autosomal recessive disorder either sex has one in four chance of being affected. The genetic locus is on chormosome 6 and despite clinical variability of ARPKD a single gene is responsible. In utero testing for ARPKD is available in advanced centers.
Final Discussion
Thank you Dr. Nandita. The diagnosis in this case was not in doubt, however, one of the
issues that merits discussion is whether the termination of pregnancy should
have
been con-
sidered despite the fact that we were not sure at that time about the distinction between ADPKD or ARPKD. The ADPKD presents with advanced renal failure at an early stage and therefore it l1eeds to be discussed.
Dr. Vanita Jain: Malformations such as PKD are picked up by ultrasound around 18th weeks of pregnancy when bladder is not visualized and the liquor is reduced indicating
negligible renal function. Whether ADPKD or ARPKD the prognosis of the baby is usually poor which is explained to the mother. The choice of terminating the pregnancy by a mid- term abortion or continuing the pregnancy is given to the mother. Terminating a pregnancy at that time has a lot of complications and usually no active intervention is advised for fetal indications either by induction of labor or by Cesarean section. The mother of the baby was also explained on the same lines and the prognosis of the baby was clearly stated. She chose to continue the pregnancy and went to preterm labor. Obstetrically it is much safer for mother to go into labor but if the mother wants termination it can be done.
Dr. V. Jha: The diagnosis of ARPKD should have serious complications. When it is picked up, a lot of counselling may be given, to the parents.
Dr. Vanita Jain: The mother of this baby has been advised to come after six weeks for counselling and we will explain to her the risk involved. In case she plans to conceive again and' take the risk she would be scanned periodically. Even if the kidneys appear
normal
at 16 weeks of gestation it does not
mean that the baby is not going to develop malformation in future.
Dr. K.S. Chugh: I think Dr. Vanita has al- ready highlighted that by the time you detect PKD the harm is already done. However, there is lot of technology coming up to detect such lesions much earlier than the ultrasound evidence. Dr. Sakhuja and Dr. Jha have recently reviewed this subject regarding its molecular basis and genetic engineering.
I would like to ask the pathologist if there is any difference in the juvenile and teenage varieties? I thought it was simply the extension of age.
Dr. Nandita Kakkar: The difference between these two varieties is the age of presentation. The juvenile from presents between 1-5 years whereas teenage variety presents during the latterpart of the second decade.
Dr. V. Sakhuja: As regards the division of ARPKD into perinatal. infantile and juvenile groups, these divisions do reflect the age of the onset of symptoms and they are not really the different forms of the disease and there is no genetic heterogeneity. DNA markers that are linked to abnormal gene on chromosome 6 can be used for antenatal diagnosis of PKD but such facilities are available only in limited
number of centers.
Dr. S. Jain: There are genetic differences between ADPKD and ARPKD. For ADPKD there are known abnormalities on chromosome 16 and chromosome 6 but despite that one may not pick up the diagnosis. The diagnosis of ARPKD
is usually based on ultrasound findings. I would also like to point that the size of kidneys in a fetus on ultrasound may be more subjective rather than accurate. A difference of 2-4 mm in size may mean a lot in the terms of prognosis in view of the fact that Dr. Vanita
has pointed out earlier that in ADPKD the size is slightly more at 16 weeks. In my view the real difference is appreciated at the start of the third trimester by which time it is not possible to offer any help in the form of termination of pregnancy.
Dr. Vanita Jain: I do not agree that antenatally it is difficult to measure the accurate size of fetal kidneys. The size of the kidney is very well defined and known for a particular gestational age and enlarged kidneys can be
well appreciated at ultrasound even if the enlargement is by more than 1-2 mm.
Dr. Jayashree: I would like to comment on the prenatal diagnosis of ARPKD and ADPKD. DNA probes are available for pre- natal diagnosis of dominant form of PKD where definite loci of PKD1 and PKDz are identified. PKDJ is also known but yet to be mapped. Such DNA probes are not available for recessive form of PKD and for its diagno- sis one has to depend mostly on ultrasound findings. If there is a past history of PKD in sibling as seen in this case it may be an important clue for the diagnosis of ARPKD alongwith ultrasound findings.
Dr. V. Sakhuja: Thank you very much. I close the discussion.
|
1.
Groupe WE. Differential diagnosis of enlarged kidneys. In: Textbook of Disease of the New- born, 5th edn. Eds. Avery ME, Taeusch HW. Philadelphia, W.B. Saunders and Company, 1984; pp 447-451.
2.
Bernstein J. Renal hypoplasia and dysplasia. In: Textbook of Pediatric Kidney Diseases, 2nd edn;- Vol II Ed. Edelmann CM, Boston, Little Brown and Company, 1992: pp 1121-1137.
3.
Bernstein J, Siovis LT. Polycystic disease pf kidney. In: Textbook of Pediatric Kidney Diseases, 2nd edn., Vol II. Ed. Edelmann CM, Bos- ton, Little Brown and Company, 1992; pp 1139-1157.
4.
Kaplan BS, Kaplan P, Rosenberg HK, Lamothe E, Rosenblatt DS. Polycystic kidney diseases in childhood. J Pediatr 1989; 115: 867- 880.
5.
Cole BR. Conley BS, Stapleton BF. Polycystic kidney disease in the first year of life. J Pediatr 1987; 1 II: 693-699.
6.
Romero R, Cullen M, Jeanty P, Grannium P, Reece EA, Venus I, et at. The diagnosis of congenital renal anomalies with ultrasound. Am J Obstet Gynecol1984; 150: 259-262.
7.
Weil LB, et al. Unilateral neonatal cystic disease of the kidney as first manifestation of tube- rose sclerosis. Arch Esp Urol 1997; 50: 1012- 1014.
8.
Mishra S, Kumari S, Niranjan S, Sachdev CP, Bajaj P. Cystic kidney disease. Indian Pediatr 1996; 33: 134-140.
|