O
steoporosis pseudoglioma
syndrome (OPPGS) is a rare autosomal recessive genetic disorder
characterised by congenital- or infantile-onset blindness and severe
osteoporosis [1,2]. It is caused by loss-of-function biallelic mutations
in the lipoprotein receptor-related protein 5 (LRP5) gene.
We describe a family from southern India with four members affected with
OPPGS, wherein a novel homozygous missense pathogenic variant was
identified in the LRP5 gene. Significant variability in the
phenotypic severity was noted in the affected family members, which is
unusual for an autosomal recessive genetic disorder. Initiation of
bisphosphonate therapy resulted in significant clinical improvement in
the two severely affected members over a 6-month follow-up period.
Case Report
The index patient (IV.3), a 4-year-old female child,
the third offspring of 3
rd
degree consanguineous parents (Fig. 1) who presented with
a referral diagnosis of osteogenesis imperfecta, had history of multiple
fractures (five fractures involving bilateral lower limbs and right
upper limb) since early infancy and complete blindness of both eyes
since birth. Her antenatal and perinatal periods had been uneventful.
Though developmentally normal in all other spheres, she had attained the
ability to stand with support by 3 years but had never been able to
walk, even with support. There was no history of hearing loss, other
focal neurological deficits or any other systemic symptoms.
On examination, the proband’s anthropometric
measurements were as follows: height 90 cm (5
th
centile), weight 10 kg (-2 to-3 SD) and head circumference 43 cm (-4 to
-5 SD). She had bilateral microcornea with corneal opacities, with white
sclerae. The craniofacial dysmorphic features noted included
microcephaly, enophthalmos, mildly low set ears, prominent zygomatic
arches and bulbous tip of nose (Web Fig. 1).
Bowing of both thighs and legs was present. The neurological examination
was normal except for complete loss of vision in both eyes and mild hypotonia in both the lower limbs. No abnormality was found on
examination of the cardiovascular, respiratory and abdominal systems.
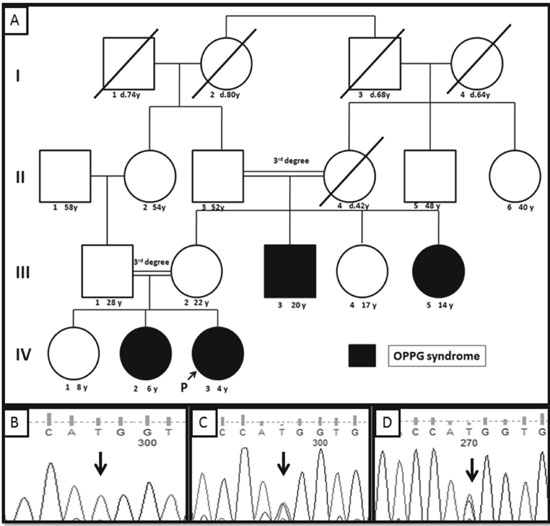 |
|
Fig. 1 (a)Pedigree of the
reported family; (b) Sanger sequence chromatogram of the
proband showing homozygous c.3709C>T (marked by black arrow)
variant in the LRP5 gene; (c & d) Sanger sequence chromatograms
of the proband’s father and mother respectively showing the
heterozygous c.3709C>T (marked by black arrow) variant in the
LRP5 gene.
|
A detailed family history was elicited which revealed
similar symptoms in one maternal uncle (III.3), one maternal aunt
(III.5) and one elder female sibling (IV.2) as indicated in the pedigree
in Fig.1(a) and in Table I.
Examination of these other three affected family members revealed
similar findings, but with variable severity. The uncle (III.3) had
short stature (height 145 cm; -3 to -4 SD), normal head circumference
(54.5 cm), bilateral microphthalmia with corneal opacification, no other
significant facial dysmorphism, widely spaced and eroded teeth, swollen
and tender knee joints and bilateral anteriorly bowed tibiae. The
affected aunt (III.5) had a normal height (143 cm; 5th
centile) with no skeletal deformity, microcephaly (head circumference
50.5 cm; -2 to -3 SD), bilateral microcornea with corneal opacities,
enophthalmos, low-set ears and bulbous tip of nose. The affected sister
(IV.2) had short stature (height 101 cm; -2 to -3SD), microcephaly (head
circumference 46 cm; -3 SD), the same eye and facial dysmorphic findings
as the proband, and no skeletal deformities.
TABLE I Clinical Features in the Affected Members of the Reported Family Demonstrating Variable Expressivity of the Disease
|
Clinical features |
Affected uncle (III.3) |
Affected aunt (III.5) |
Affected sister (IV.2) |
Proband (IV.3) |
|
Fractures
|
+ (5 episodes) |
+ (1 episode) |
-
|
+ (5 episodes) |
|
Age at 1st fracture
|
9 years |
12 years
|
-
|
Infancy
|
|
Blindness
|
Complete
|
Partial vision in one eye
|
Complete
|
Complete
|
|
Motor developmental delay
|
-
|
-
|
+
|
+
|
|
Microcephaly
|
-
|
+
|
+
|
+
|
|
Short stature
|
+
|
-
|
+
|
- |
|
Hearing loss
|
+
|
-
|
-
|
-
|
|
Vertebral compression |
- |
- |
- |
+ |
|
Dysmorphic features |
|
|
|
|
|
Deep set eyes |
+
|
+
|
+
|
+
|
|
Asymmetric palpebral fissures |
- |
- |
- |
+ |
|
Sparse lateral third of eyebrows |
+
|
+
|
+
|
- |
|
Prominent zygomatic arches |
+
|
+
|
+
|
+
|
|
Bony prominence over lateral
|
- |
- |
+ |
- |
|
supra orbital margin |
|
|
|
|
|
Bulbous nasal tip |
+
|
+
|
+
|
+
|
|
Thick lips |
+
|
+
|
+
|
+
|
Radiographs of the spine, pelvis, femur and knee in
the proband and the other three affected family members were suggestive
of diffuse osteopenia. The proband additionally had vertebral
compression (Web Fig. 1). Serum calcium, phosphorous,
alkaline phosphatase and total vitamin D3 were within normal limits.
Ophthalmological examination revealed evidence of foveal exudative
vitreoretinopathy with phthisis bulbi in all four individuals.
Based on the above findings, a provisional diagnosis
of Osteoporosis pseudoglioma syndrome was made and sequencing of the
LRP5 gene was done. A novel homozygous c.3709C>T (p.Arg1237Trp)
missense sequence variant was identified in the proband in exon 17 of
the LRP5 gene (Fig.1b) through targeted gene
panel testing using the Illumina sequencing platform (Illumina Inc., San
Diego, California, United States) and further validated through Sanger
sequencing using the ABI 3130 automated genetic analyzer (Life
Technologies, Thermo Fisher Scientific Corporation, Foster City,
California, USA). This sequence variant was not present in the dbSNP,
1000 Genome and Exome Aggregation Consortium (ExAC) databases. The
pathogenicity of this variant was inferred based on the results of the
variant prediction software Polyphen-2, Mutation Taster, and SIFT.
Targeted mutation analysis of the other family members was done through
Sanger sequencing; the affected uncle (III.3), affected aunt (III.5) and
elder sister (IV.2) of the proband were confirmed to be homozygous for
the c.3709C>T variant and the parents (III.1 and III.2) and maternal
grandfather (II.3) were confirmed to be heterozygous carriers for the
same (Fig. 1c and 1d).
The proband and her affected elder sister were
started on intravenous Pamidronate therapy at a dose of 1mg per kg body
weight for three consecutive days, once every 3 months and the affected
uncle and aunt were started on oral bisphosphonate therapy (oral
Alendronate in the dose of 1mg per kg body weight once a week) with
daily calcium supplementation. The proband was followed up after 3
months and 6 months, by which time she showed remarkable clinical
improvement with no fractures reported during the period and had
attained the ability to walk with one hand held over a distance of up to
2 metres. The severely affected uncle also reported a marked clinical
improvement with cessation of pain, ability to stand by himself without
support and ability to walk with the help of a walking stick.
Discussion
The LRP5 protein is believed to play a role in
determining bone mineral density through the Wnt signalling pathway and
in retinal vascularisation through Norrin/Frizzled 4 signalling [3-6].
Loss-of-function mutations in the LRP5 gene are therefore
associated with osteoporosis and exudative vitreoretinopathy. Variable
expressivity and intra-familial variability, though usually associated
with autosomal dominant disorders, may occur with autosomal recessive
conditions. There was marked intra-familial variability of the OPPGS
disease-phenotype in the reported family.
Many short-term studies on bisphosphonate therapy in
OPPGS have reported beneficial effects, which include reduction in bone
pains, increased bone mineral density, decreased fracture rate and
subsequent improvement in the quality of life [7,8]. However, long-term
follow-up data pertaining to the use of bisphosphonates in OPPGS is
limited. A recent study [9] reported that though there is significant
improvement in the areal bone mineral density (aBMD) in
bisphosphonate-treated OPPGS patients, the trabecular volumetric BMD
(vBMD) remains low and therefore in the long term there is no
significant improvement in the bone fragility.
Further studies in such cases to look for causes of
variable expressivity may throw a light on novel disease/phenotype
modifying genes and genomic variants. This case also highlights the
importance of screening for osteopenia in cases of familial exudative
vitreoretinopathy for early intervention.
Contributors: KBT, PR: clinical evaluation and
diagnosis of patient, review of literature, preparation of manuscript;
ABD: genetic evaluation of patient, preparation and review of
manuscript.
Funding: Funding support for molecular genetic
evaluation of patients from core funds of Centre for DNA Fingerprinting
and Diagnostics, Hyderabad.
Competing Interests: None stated.
References
1. Bianchine JW, Briard-Guillemot ML, Maroteaux
P, Frezal J, Harrison HE. Generalized osteoporosis with bilateral
pseudoglioma—an autosomal recessive disorder of connective tissue:
Report of three families—review of the literature. Am J Hum Genet.
1972;24:34A.
2. Beighton P, Winship I, Behari D. The ocular
form of osteogenesis imperfecta: a new autosomal recessive
syndrome. Clin Genet. 1985;28:69-75.
3. Shaharao V, Shah I, Mishra P, Muranjan
M, Bharucha B. Osteoporosis pseudoglioma syndrome. Indian
Pediatr. 1999;36:313-5.
4. Wang Y, Rattner A, Zhou Y, Williams J,
Smallwood PM, Nathans J. Norrin/Frizzled4 signaling in retinal
vascular development and blood brain barrier plasticity. Cell.
2012;151:1332-44.
5. Gilmour DF. Familial exudative
vitreoretinopathy and related retinopathies. Eye. 2015;29:1-14.
6. Gong Y, Slee RB, Fukai N, Rawadi
G, Roman-Roman S, Reginato AM, et al. LDL receptor-related
protein 5 (LRP5) affects bone accrual and eye development. Cell.
2001;107:513-23.
7. Streeten EA, McBride D, Puffenberger E,
Hoffman ME, Pollin TI, Donnelly P, Morton H.
Osteoporosis-pseudoglioma syndrome: Description of 9 new cases and
beneficial response to bisphosphonates. Bone. 2008;43:584-90.
8. Barros ER, Dias da Silva MR, Kunii IS,
Lazaretti-Castro M. Three years follow-up of pamidronate therapy in
two brothers with osteoporosis-pseudoglioma syndrome (OPPG) carrying
an LRP5 variant. J Pediatr Endocrinol Metab. 2008;21:811-8.
9. Streeten EA, Ramirez S, Eliades M, Jaimungal S, Chandrasekaran S,
Kathleen R, et al. Fractures on bisphosphonates in osteoporosis
pseudoglioma syndrome (OPPG): pQCT shows poor bone density and
structure. Bone. 2015;77:17-23.


