Cardiac channelopathies are a group of inherited
arrhythmias caused by mutations in the cardiac ion channels. Long QT
syndrome (LQTS), the commonest of these disorders, is estimated to
affect 1 in 2000 people [1]. The other disorders include Brugada
syndrome (BrS), catecholaminergic polymorphic ventricular tachycardia
(CPVT) and the short QT syndrome (SQTS). They share a predilection for
malignant ventricular arrhythmias, syncope and sudden death. Seizure is
a frequent presentation in these disorders and is attributed to
arrhythmia related decrease in cardiac output [2]. It is not uncommon
for children with these disorders to be inaccurately diagnosed to have
epilepsy. We report four children with cardiac channelopathies referred
from neurology in whom appropriate therapy resulted in a significant
decrease in paroxysmal ‘seizure-like’ events.
Case 1: A 5-year-old boy was referred to
us with multiple seizures after electrocardiographic (ECG) abnormalities
were noted during an extended electroencephalogram recording. A younger
sibling had multiple episodes of seizures and died at 1 year. There was
a history of sudden unexpected death in the family with one drowning. A
standard ECG in the present case showed a prolonged QTc (520 ms). Holter
evaluation documented multiple self-terminating episodes of Torsades de
Pointes (TdP). A provisional diagnosis of LQTS was made. He was started
on beta-blockers. The family were unwilling for family screening as well
as genetic testing. On follow-up, 6 months later, there were no further
symptoms.
Case 2: A 6-year-old girl, the
first child of non-consanguineous parents, had a maternal aunt with
epilepsy, high pitched sounds being one of the triggers. The child had
been diagnosed prenatally to have complete heart block and a pacemaker
had been implanted postnatally. She also had multiple episodes of
seizures. Three EEGs were reported to be normal. A review of her
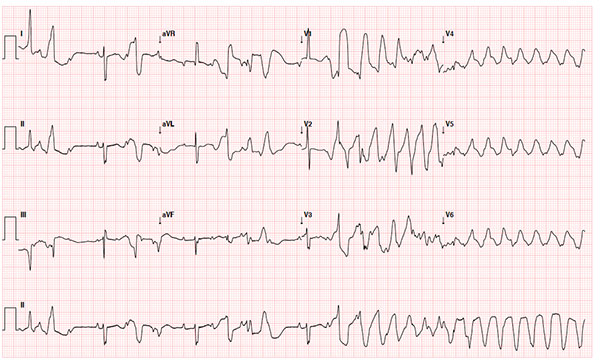
neonatal ECG showed one episode of TdP (arrow in Fig. 1a). On
family screening, the mother’s ECG showed a prolonged QTc of 480 ms.
Anti-epileptic drugs were discontinued, and she was started on beta
blockers. Genetic testing revealed a pathogenic mutation in KCNH2
gene (c.1882G>A), which causes LQTS type 2. Cascade screening confirmed
the mutation in both the mother and maternal aunt, and both were started
on beta blockers. At follow up of one year, there were no further
seizures in the child.
 |
|
Fig. 1 (a) ECG of patient 2 in the newborn period
demonstrating polymorphic ventricular ectopy as well as an
episode of Torsades de Pointes (TdP).
|
Case 3: A 17-year-old girl, the first child of
third-degree consanguineous parents, had recurrent seizures during
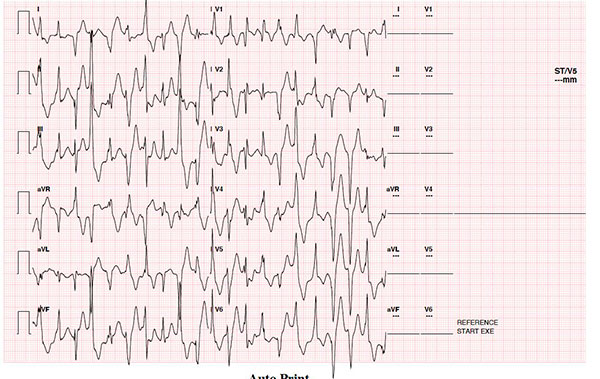
exertion. Her ECG was normal. There was bidirectional ventricular ectopy
on exercise stress testing (arrow in Fig. 1b). A clinical
diagnosis of CPVT was made and she was started on beta blockers. Genetic
testing revealed a pathogenic heterozygous mutation in the Ryanodine
receptor gene (RyR) associated with CPVT (c.184 C>T). The family
refused cascade screening. No further seizures were reported during a
follow up period of 6 months.
 |
|
Fig. 1(b) ECG tracing from an
exercise stress test of patient 3 demonstrating evidence of
bidirectional ventricular ectopy.
|
Case 4: A 9-year-old girl, the
first child of non-consanguineous parents, with two younger siblings who
had died suddenly and unexpectedly in infancy and multiple sudden
unexpected deaths in the father’s family was referred for evaluation.
She had been treated for seizures from the age of five. A prolonged
video-EEG recording was performed. She had one episode of tonic
posturing during the recording with normal EEG and TdP on ECG. Baseline
ECG showed sinus rhythm with a normal QTc. A presumptive diagnosis of
LQTS was made, she was started on beta blocker and genetic testing was
organized. There have been no further episodes of seizures for the last
9 months.
Cardiac channelopathies are caused by mutations in
proteins related to the intra-cellular transport of sodium, potassium
and calcium ions. These ion channel abnormalities predispose the patient
to episodes of lethal ventricular arrhythmias such as TdP and
ventricular tachycardia (VT) or fibrillation (VF). While some may die
during such arrhythmias, these lethal arrhythmias can be non-sustained
with spontaneous termination. Cardiac output is significantly decreased
during these non-sustained episodes and the resultant cerebral
hypoperfusion may result in seizures and/or syncope [3]. The term
‘torsadogenic seizures’ has been used to describe this kind of
paroxysmal activity in the past [4]. Misdiagnosis as epilepsy has been
shown to be the most important reason for delay in diagnosis and could
potentially result in sudden death [5].
A careful history can provide clues to the diagnosis
in most patients. A family history of SUD, seizures or syncope in
multiple members should raise the suspicion of a cardiac channelopathy
[6], as was seen in three of our cases. Recurrent seizures despite
appropriate therapy and especially in the absence of pathogenic EEG
changes should raise the suspicion of a cardiac channelopathy [6].
Genetic testing by a targeted panel of genes
implicated in cardiac channelopathies or an exome wide screening is
performed through next generation sequencing. A positive genetic test
allows genotype specific therapy [7]. Genetic testing also permits
cascade testing in other (even asymptomatic) family members [8] and
helps identify individuals at risk and provide appropriate treatment
even before clinical manifestations occur. This is especially important
in channelopathies as a normal ECG does not rule out the presence of the
phenotype. The QTc interval may be normal on a baseline ECG in affected
individuals and an exercise stress test or provocative testing with
adrenaline may be necessary to identify QTc prolongation. In our series,
cascade screening permitted diagnosis in an asymptomatic mother as well
as an aunt in whom the symptoms had wrongly been attributed to seizures.
In conclusion, seizures may be the clinical
presentation in children with cardiac channelopathy. Red flags such as a
family history of sudden death, seizures associated with auditory
triggers and recurrent seizures with a normal EEG should raise suspicion
and prompt referral to a pediatric cardiologist.The institution of
appropriate lifestyle modifications and pharmacological therapy could
result in control of symptoms.
REFERENCES
1. Modi S, Krahn AD. Sudden cardiac arrest
without overt heart disease. Circulation. 2011;123:2994-3008.
2. Anderson JH, Bos JM, Cascino GD, Ackerman MJ.
Prevalence and spectrum of electroencephalogram-identified epilepti-form
activity among patients with long QT syndrome. Heart Rhythm.
2014;11:53-7.
3. Sun AY, Pitt GS. Long QT syndrome and
seizures. JACC Clin Electrophysiol. 2016;2:277-8.
4. Johnson JN, Hofman N, Haglund CM, et al.
Identification of a possible pathogenic link between congenital long
QT syndrome and epilepsy. Neurology. 2009;72:224-31.
5. MacCormick JM, McAlister H, Crawford J, et al.
Misdiagnosis of long QT syndrome as epilepsy at first presentation.
Ann Emerg Med. 2009;54:26-32.
6. Hazle MA, Shellhaas RA, Bradley DJ, Dick M,
Lapage MJ. Arrhythmogenic channelopathy syndromes presenting as
refractory epilepsy. Pediatr Neurol. 2013;49:134-37.
7. Mazzanti A, Maragna R, Faragli A, et al.
Gene-specific therapy with mexiletine reduces arrhythmic events in
patients with long QT syndrome type 3. J Am Coll Cardiol.
2016;67:1053-8.
8. Sturm AC. Cardiovascular cascade genetic testing: Exploring the
role of direct contact and technology. Front Cardiovasc Med. 2016;3:11.


