|
reminiscences from Indian Pediatrics: A tale
of 50 years |
|
|
Indian Pediatr 2015;52:
795-801 |
 |
Hereditary Multiple
Exostoses – A Tale of 50 years
|
|
Preeti Singh and *Sharmila
B Mukherjee
Department of Pediatrics, Lady Hardinge Medical College, New Delhi,
India.
*Email: [email protected]
|
|
 We continue our expedition into the past while
reviewing an article from the September 1965 issue of Indian Pediatrics.
This comprised of 41 pages with four research papers (ECG changes in
progressive muscular dystrophy, a mathematical method to estimate gene
frequencies, antenatal illnesses in villages of Punjab and a case
series), two case records (pulmonary agenesis, polyarteritis nodosa),
book reviews, current literature and news. We selected the case series
of Hereditary multiple exostoses (HME), primarily to showcase the
meticulousness with which the authors attempted to trace back the
pedigree [1]. After briefly discussing the scientific knowledge that
existed then, we will touch upon the recent advances that have evolved
in understanding this disorder at the molecular and genetic levels. We continue our expedition into the past while
reviewing an article from the September 1965 issue of Indian Pediatrics.
This comprised of 41 pages with four research papers (ECG changes in
progressive muscular dystrophy, a mathematical method to estimate gene
frequencies, antenatal illnesses in villages of Punjab and a case
series), two case records (pulmonary agenesis, polyarteritis nodosa),
book reviews, current literature and news. We selected the case series
of Hereditary multiple exostoses (HME), primarily to showcase the
meticulousness with which the authors attempted to trace back the
pedigree [1]. After briefly discussing the scientific knowledge that
existed then, we will touch upon the recent advances that have evolved
in understanding this disorder at the molecular and genetic levels.
The Past
The study published in this issue by
Sharma, et al. [1] from King George Medical College Lucknow,
reported two affected Indian children. The first patient was an
11-year-old boy who developed multiple, hard, painless, gradually
increasing swellings over a year. At presentation, lower ends of both
femurs, tibiae and radii, upper ends of humerii and 6th
/7th ribs were involved. There was no significant
family history (traced till the second generation). The second patient
was an unrelated 12-year-old boy with similar swellings since the age of
six years. Medical attention was sought only when an injury lead to a
swelling on his left arm becoming tender and fungating. On elicitation
of details, a strong family history was unearthed (probably accounting
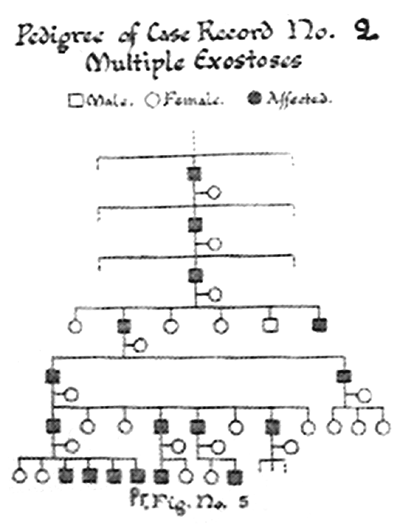
for the apparent indifference exhibited prior to injury). Pedigree
analysis (Fig. 1) revealed 17 asymptomatic (except
cosmetic) males belonging to 7 preceding generations with a striking
absence of disease in females. This was not discussed by the authors but
it may simply reflect missing data due to less severe forms associated
with females or the gender bias in healthcare seeking behavior.
In both of the cases, calcium, phosphorus and serum alkaline phosphatase
levels in serum were normal. The only salient diagnostic findings were
radiological (described later). Biopsies demonstrated proliferative
chondroid tissue. The fungating mass in the second case was infectious,
not malignant. The discussion of this article dwelt on various
nomenclature, demographic characteristics, hereditary pattern (64%
familial, remaining unknown or due to a new mutation), and clinical and
radiological features (bony projections of various size and contour with
the apex pointing towards the shaft and away from the nearest epiphysis,
heterogeneously radio-opaque with interspersed areas of translucency).
 |
|
Fig. 1 Pedigree chart of case 2 as
published in the original case record in September 1965. (Note
absence of involvement of female family members).
|
Historical background and past knowledge: A
patient with multiple exostoses was presented in a lecture by Hunteras
as early as 1786, while another with familial involvement was reported
in 1814 [2,3]. Various terms based on the typical clinical and
histopathological features (i.e. multiple osteochondromas,
cartilaginous exostosis, deforming chondrodysplasia, diaphyseal aclasis)
were in use. The prefix ‘hereditary’was added once the familial nature
was recognized. HME remained an enigma till the 1950s when some
groundbreaking research emerged. Solomon, et al. [4] delineated
the clinical, radiological, pathological and genetic characteristics of
HME in 1963. In this paper, it was stated that exostoses were
cartilage-capped bony outgrowths from the juxta-ephipyseal regions of
rapidly growing ends of bones originating in cartilage. Long bones and
less commonly flat bones were described to be involved with sparing of
face and skull. It was described that lesions appear by the end of the
first decade, increase during puberty, and become dormant with cessation
of growth. The number, size and location of exostoses were reported to
vary between and within families.
The Present
The term ‘Multiple osteochondromatosis (MO)’ adopted
by World Health Organization is preferentially used as it accurately
denotes that the lesions are cartilaginous processes that ossify, rather
than bony outgrowths (as ‘exostoses’ implies) [5]. Inheritance is
autosomal dominant with 100% penetrance in males and 96% in females
[6,7]. Sporadic de-novo mutations account for the 10% of cases in which
family history is negative. Diagnosis is established by the presence of ³2 typical
radiological lesions with or without positive family history [5].
There are two variants of MO with
genotypic-phenotypic correlation. Type I due to mutations in tumor
suppressor genes EXT1 (exostosin-1) on chromosome 8q23-24.1 is
more severe, commonly involves flat bones, and is associated with
shortened stature and malignant transformation [8]. Type II is due to
mutations in the EXT2 (exostosin-2) genes on chromosome 11p11-13
[9]. Both these genes encode glycosyl transferase necessary for
biosynthesis of heparan sulfate in many cells, including chondrocytes.
In the latter, proteoglycans are secreted into the extra-cellular matrix
during endochondral ossification within the growth plate. Mutations
result in truncated gene products interfering with normal chondrocyte
proliferation and differentiation, abnormal bone growth and development
of exostoses [10]. These are detected by Polymerase Chain Reaction (PCR)
and/or Multiplex ligation-dependent probe amplification (MLPA) for
definitive and prenatal/pre-implant diagnosis.
References
1. Sharma NL, Singh RN, Anand JS. Hereditary multiple
exostoses (ecchondosisossificans): 17 cases in 7 generations. Indian
Pediatr.1965;2:336-41.
2. Hunter J. In: Palmer JF, editor. The works
of John Hunter, F.R.S. Vol 1. London: Longman, Rees, Orne, Brown, Green,
and Longman; 1835.
3. Boyer A. Traité de maladies chirurgicales vol
3. Paris: Ve Migneret; 1814. p. 594.
4. Solomon L. Hereditary multiple exostosis. J Bone
joint Surg (Br). 1963;45:292-304.
5. Bovée JVMG, Hogendoorn PCW. Multiple
osteochondromas. In: Fletcher CDM, Unni KK, Mertens F,
editors. World Health Organization. Classification of Tumours. Pathology
and Genetics of Tumours of Soft Tissue and Bone. Lyon, France: IARC
Press; 2002. p. 360-2.
6. Schmale GA, Conrad EU, Raskind WH. The natural
history of hereditary multiple exostoses. J Bone Joint Surg
[Am]. 1994;76-A:986-92.
7. Legeai-Mallet L, Munnich A, Maroteaux P, Le Merrer
M. Incomplete penetrance and expressivity skewing in hereditary multiple
exostoses. Clin Genet. 1997;52:12-16.
8. Cook A, Raskind W, Blanton SH, Pauli RM, Gregg RG,
Francomano CA, et al. Genetic heterogeneity in families with
hereditary multiple exostoses. Am J Hum Genet. 1993;53:71-9.
9. Wu YQ, Heutink P, de Vries BB, Sandkuijl LA, van
den Ouweland AM, Niermeijer MF, et al. Assignment of a second
locus for multiple exostoses to the pericentromeric region of
chromosome.Hum Mol Genet. 1994;3:167-71.
10. Zak BM, Crawford BE, Esko JD. Hereditary multiple
exostoses and heparan sulfate polymerization. Biochim Biophys Acta.
2002;1573:346-55.
|
|
|
 |
|
