|
|
Case Reports Indian Pediatrics 2000;37: 1122-1125 |
|||||||||||||||||||
|
Myotonia Congenita: Response to Carbamazepine |
|||||||||||||||||||
|
Sheela S.R.
Myotonia is the inability of a muscle to relax instantly after a contraction. In myotonia congenita the persistent muscle contraction results in muscular hypertrophy. Myotonia congenita affecting twenty persons in a family over five generations is reported. Six members with ages ranging from 5 to 58 years were regularly followed up for 18 months. Two of them were willing to take treatment with Carbamazepine, and showed marked reduction of myotonia.
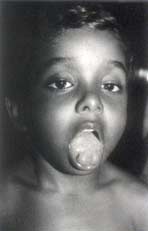
The proband was a 5-year-old boy who presented with history of difficulty in getting up from bed in the morning, and in standing up from the sitting posture, and in initating walking after sitting for some time. This "stiffness of the muscles" was greater in the early morning, during cold weather, and with anxiety; it disappeared with activity. Parents noticed these symptoms from early infancy. He was born of a non-consanguineous marriage. The pregnancy, delivery and postnatal periods were uneventful. Initially there was a mild delay in gross motor milestones, but other milestones were normal. The child weighed 19 kg and his height was 104 cm. There was marked hypertrophy of the biceps, triceps, calf muscles, and masseter, simulating an "Infant Hercules". Release of the hand grip was very slow, and there was prominent percussion myotonia of the tongue (Fig. 1). He had severe difficulty in climbing up stairs. He could not immediately open his eyes after sneezing. Muscle power, tone and tendon reflexes were normal.
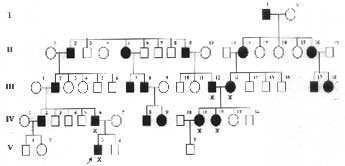
A detailed pedigree charting showed that five generations of the family had similar symptoms with an autosomal dominant inheritance pattern (Fig. 2). Six patients were examined in detail, of whom the proband, his father, and a 17-year-old girl were severely affected. The child’s father had typical signs, including generalized muscular hypertrophy, notably of the calf muscles, and percussion myotonia of the tongue. The girl too had generalized hypertrophy of muscles and percussion myotonia of the tongue and had severe myotonia of orbicularis oculi muscles after sneezing, manifesting since two years of age, and she sustained frequent falls while walking. Her symptoms were greater in the early morning, during cold weather, and with anxiety; it disappeared with activity. Her parents had mild myotonia of the tongue and they claimed that the stiffness had reduced with age.
The proband and the 17-year-old girl were investigated. The serum sodium, potassium and CPK were normal in both. EMG of both children showed the typical repetitive discharges of varying amplitude and frequency (Dive-Bomber pattern) which is the actual demonstration of myotonia. Nerve conduction velocity was normal in both. Muscle biopsy was not done as both of them had all classical features of Myotonia congenita. It was decided to treat the proband and the girl; the other patients were reluctant to undergo continuous durg therapy. Carbamazepine was started in a dose of 5 mg/kg, and gradually built up to 25 mg/kg/day. After two months there was amelioration of eyelid myotonia after sneezing. Now after 18 months of therapy these children have no difficulty getting out of bed in the morning; the difficulty in releasing grip and in initiating walking is mild; the girl no longer falls while walking; but the difficulty in climbing stairs persists. They are continuing on the drug, and no undesirable effects have been noticed.
Mytonia congenita (Thomsen disease) was first described by Thomsen in 1876 in his own family. Myotonia is persistent contraction of skeletal muscles following stimulation leading to generalized mucsular hypertrophy and ‘Herculean appearance’, but these large muscles are weak. Myotonia involves most of the voluntary muscles but hypertrophy is principally seen in the limbs and trunk. Tongue, face and jaw muscles can also be involved. Persistent muscle contraction results in muscular hypertrophy. The muscle stiffness is painless, and is exacerbated by anxiety, cold and fatigue. Percussion myotonia is prominent. Myotonia usually develops by 2-3 years of age. This autosomal dominant disease should be differentiated from the rare recessive generalized myotonia (Becker type myotonia) which is similar, but has a later age of onset, a more severe nature, and manifests transient muscle weekness during muscle exertion after rest(1). Positive family history, early appearance and lack of progression of myotonia and presence of generalized muscular hypertorphy distinguish myotonia congenita from myotonic dystrophy. Paramyotonia congenita is a myotonia that is precipitated by cold. Myotonic chondrodystrophy (Schwartz-Jampel disease) has myotonia, dysmorphic phenotypical features, dwarfism and joint abnormalities(2). Myotonia congenita is bascially a chloride channel myopathy. CLCN1, the gene encoding the major skeletal muscle chloride channel, is localized to chromosome 7q35 locus. A mutation in this gene can cause either dominant Thomsen or recessive Becker type of myotonia(3). Diagnosis is established by EMG, which shows the waxing and waning amplitude and frequency of potentials, producing the characteristic sound (Dive-Bomber pattern) due to myotonic discharges. CPK and nerve conduction velocities are normal. Muscle biopsy specimens in both the dominant and recessive type demonstrate absence of type II b fibers. Both the patients in this report showed remarkable improvement with carbamazepine, which can be safely used for a long duration. Though phenytoin has shown better results in earlier studies, due to the undesirable side effects such as hirsutism, gingival hyper-trophy, and blood dyscrasias, it was avoided. Myotonia can be controlled by drugs that raise the depolarization threshold of muscle membranes, e.g., carbamazepine, phenytoin, procainamide and quinidine. Carbamazepine and phenytoin are given in usual anti-convulsant doses(2). Among the antiarrythmic drugs, mexiletine and tocainide are the most antimyotonic agents acting by their fast blocking effect on voltage dependant sodium channels in the muscle membranes. Because of the risks of hematological problems, tocainide is not recommended for the treatment of myotonia(4). Acetazolamide is useful in myotonia, but long term treatment can cause nephrolithiasis(5). In one study from Portugal, patients were treated with different drugs to determine the therapy for optimal relief of myotinia. Five patients responded to phenytoin, one to carbamazepine, three to acetazolamide and none to quinidine or procainamide(6). Another double blind study compared phenytoin and carbamazepine on myotonic patients; there was a good response to both drugs, but phenytoin showed a decreased efficiency at higher doses(7). Mexiletine has shown excellent results in an infant with early-onset myotonia congenita(8). Dantrolene may be tried in patients who do not respond to the usual drugs(9). Myotonic patients react unfavourably to many anesthetic drugs and sedatives. Succinyl choline is contraindicated as it can cause prolonged contraction, and difficulty in intubating and ventilating the patient; and increased release of potassium which can precipitate an arrhythmia. Spinal and epidural anesthesia are safer than general anesthesia. These patients also have a higher risk of pulmonary aspiration and post-operative pneumonia(10). If they need to undergo surgery, the anesthesiologist should be alerted to the diagnosis. Contributor: SSR is the sole author who has treated these patients and drafted the paper. She will act as the guarantor for the paper. Funding:
None.
| |||||||||||||||||||
![]()