|
|
|
Indian Pediatr 2021;58: 1096-1097 |
 |
Congenital Diarrheal Disorders in Neonates: A
Single-Center Experience
|
|
Shyam Sundar Sharma,1 Srinivas Sankaranarayanan,2
Vaanathi Hementha Kumar,1* Natarajan Chandra Kumar,1 C Shanmuga
Sundaram1
From 1Departments of Neonatology and 2Pediatric
Gastroenterology,
Kanchi Kamakoti CHILDS Trust Hospital, Chennai.
Email:
[email protected]
|
|
Congenital diarrheal disorders (CDDs) are a group of inherited diarrheas
with typical onset in the early neonatal period [1,2], with most being
gene specific [3]. There is limited literature on the clinical spectrum
and outcome of CDDs from India. Molecular genetic analysis has become
the preferred diagnostic modality in recent years [4,5]. Here we report
a case series of six cases of neonatal-onset chronic diarrhea (>14 days)
with their outcomes over a 3-year period (2017- 2020) (Table I)
[6].
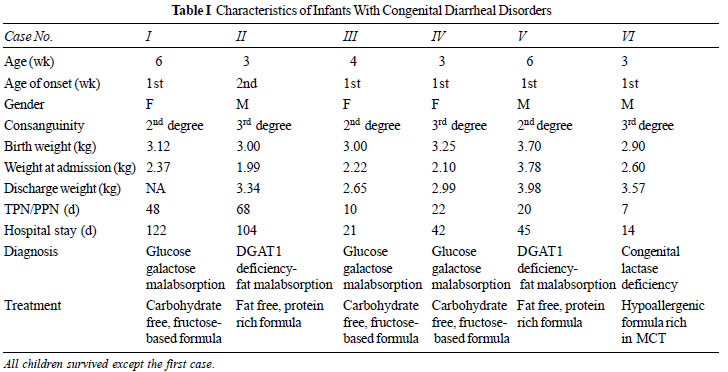 |
Cases No. 1, 3 and 4: These patients were
diagnosed with congenital glucose-galactose malabsorption (CGGM). All
three developed diarrhea on exclusive breast feeds during the first week
of life. Case No. 1 presented with osmotic diarrhea, hypoglycemia,
hypernatremia and metabolic acidosis and septicemia. There was no
evidence of immune deficiency. There was no improvement on
hypoallergenic formula and total parenteral nutrition (TPN). The child
eventually succumbed to fungal sepsis without a diagnosis. Next
generation sequencing (NGS) sent 3 weeks prior to death provided the
diagnosis of CGGM posthumously. Parents were advised prenatal counseling
for the next pregnancy. Case No. 3 and 4 with CGGM had a very similar
presentation. Oral rehydration solution (ORS) glucose challenge [8] was
positive in both the cases. Endoscopic biopsy and electron microscopy
were unremarkable. In view of history of consanguinity, lack of response
to amino acid-based formula (AAF) and a clinical picture that resembled
CGGM, they were commenced empirically on fructose-based special formula
(FBF) pending the final reports of NGS (Next generation sequencing).
There was dramatic clinical recovery with complete resolution of
diarrhea within 48 hours and TPN was discontinued. Optimal and
consistent weight gain was achieved prior to discharge. Molecular
genetic analysis by NGS confirmed the diagnosis during follow-up. Both
children, when last assessed at 2 years of age, were found to be
thriving well and had achieved age-appropriate developmental and social
milestones.
Case No. 2 and 5: These two patients were
diagnosed with diacylglycerol acyltransferase (DGAT-1) deficiency. They
presented during the second week of life with feed refusal and failure
to thrive on exclusive breastfeeds. Formula feed supplementation also
resulted in vomiting, dehydrating diarrhea and hypoalbuminemia.
Continuous nasogastric infusion of AAF did not resolve the symptoms.
Investigations did not reveal any evidence of sepsis or immune
deficiency. Oral glucose challenge was negative. Endoscopic biopsies
appeared to show nonspecific patchy villous atrophy with no viral
inclusion bodies. Electron microscopy was normal. They were started on
TPN while awaiting a genetic diagnosis. NGS confirmed DGAT-1 deficiency
and they were treated with a special custom-made fat free infant
formula, fat soluble vitamins and MCT oil. These children, in addition
to dehydrating diarrhea and FTT, had recurrent vomiting, hypoalbuminemia,
hypertriglyceridemia and occasional bulky/greasy stool classical of DGAT
-1 deficiency. Case No. 2 is currently aged 18 months and has motor
developmental delay. The child continues to fail to thrive on the
fat-free specially formulated diet. Case No. 5 also responded to
fat-free diet and showed slow weight gain, but is now lost to follow-up.
Case 6: This patient presented with neonatal
cholestatic jaundice, osmotic diarrhea on exclusive breastfeeds and
failure to thrive. The jaundice disappeared gradually but diarrhea and
failure to thrive persisted despite adequate breastfeeds. The child
continued to remain symptomatic even on supplemental infant formula.
Serum total IgE levels were elevated, suggesting atopy. The child
improved dramatically on a trial of hypoallergenic formula rich in MCT.
NGS revealed an eventual diagnosis of congenital lactase deficiency. The
baby is growing well and is now able to tolerate lactose-free cow milk
protein containing infant formula at 9 months of age.
We, herein, describe the clinical spectrum of
genetically confirmed CDDs; though electron microscopy aided diagnosis
of MVID has previously been reported [9]. Our case series showed that
congenital brush border enzyme deficiencies are the most common form of
CDDs rather than congenital enteropathies or ion channelopathies. CGGM
has autosomal recessive inheritance with classical triad of
hypernatremia, hypoglycemia and metabolic acidosis [4,7]. All children
with CDDs were born of consanguinity and diarrheal onset was within the
first 2 weeks with classical triad. For DGAT-1 deficiency, literature
cites resolution of diarrhea with fat free formula and a possible need
for fat soluble vitamin supplementation and intra lipid infusions [10].
Both the neonates in our case series had resolution of diarrhea;
however, had slow weight gain on fat free formula.
NGS has revolutionized the diagnostic approach to
CDDs; however, it is expensive and turnaround time is late (4 weeks). It
is more precise and is reliable than stool microscopy and stool
electrolytes. The triad of clinical presentation, tissue electron
microscopy and NGS form the cornerstone for apt diagnosis of CDDs. The
management is individualized based on the molecular and tissue diagnosis
and ranges from simple change to specialized specific diet to complex
lifelong TPN.
Contributions: SSS,VHK: responsible for patient
management, data collection and manuscript writing; NCK,SS: responsible
for drafting the paper; NCK will act as guarantor of the study; CSS:
helped in manuscript writing. The final manuscript was approved by all
authors. Funding: None; Competing interest:
None stated.
REFERENCES
1. Guarino A, Spagnuolo M.I, Russo S, et al. Etiology
and risk factors of severe and protracted diarrhea. J Pediatr
Gastroenterol Nutr. 1995;20:173-78.
2. Berni CR, Terrin, G, Cardillo G, et al. Congenital
diarrheal disorders: Improved understanding of gene defects is leading
to advances in intestinal physiology and clinical management. J Pediatr
Gastroenterol Nutr. 2010;50:360-66.
3. Ruemmele F.M. Chronic enteropathy: Molecular
basis. Nestle Nutr Workshop Ser Pediatr Program. 2007;59:73-85.
4. Terrin G, Tomaiuolo R, Passariello A, et al.
Congenital diarrheal disorders: An updated diagnostic approach. Int J
Mol Sci. 2012;13:4168 85.
5. Thiagarajah JR, Kamin DS, Acra S, et al. Advances
in evaluation of chronic diarrhea in infants. Gastroenterology.
2018;154:2045.
6. Younis M, Rastogi R, Chugh A, et al. Congenital
diarrheal diseases. Clin Perinatol. 2020;47:301-21.
7. Anderson S, Koniaris S, Xin B, et al. Congenital
glucose–galactose malabsorption: A case report. J Pediatr Health Care.
2017;31:506 510.
8. Lee WS, Tay CG, Nazrul N, et al. A case of
neonatal diarrhoea caused by congenital glucose-galactose malabsorption.
Med J Malaysia. 2009; 64:83-5.
9. Lingaldinna S, Sundaram M, Kamalarathnam CN, et
al. Congenital fatal diarrhea in newborns. Indian J Pediatr.
2017;84:953-54.
10. Van Rijn JM, Ardy RC, Kuloðlu Z, et al. Intestinal failure and
aberrant lipid metabolism in patients with DGAT1 deficiency.
Gastroenterology. 2018;155:130-43.
|
|
|
 |
|
