|
|
|
Indian Pediatr 2010;47:
277-279 |
 |
Supernumerary Marker Chromosome in a Child
with Microcephaly and Mental Retardation |
|
Frenny Sheth, Joris Andrieux* and Jayesh Sheth
From FRIGE (Foundation for Research in Genetics and
Endocrinology), Institute of Human Genetics, FRIGE House, Jodhpur Road,
Satellite, Ahmedabad 380 015, India; and *Laboratory of Medical Genetics,
Jeanne de Flandre Hospital, CHRU de Lille, 59037 Lille Cedex, France.
Correspondence to: (Foundation for Research in Genetics
and Endocrinology), Institute of Human Genetics, FRIGE House, Jodhpur
Road, Satellite, Ahmedabad 380 015, India.
Received: December, 1, 2008;
Initial review: December, 12, 2008;
Accepted: February 19, 2009.
|
|
Abstract
A de novo supernumerary marker chromosome
(SMC) was identified in a 13-month-old girl who presented with
microcephaly and mild mental retardation. On further characterization by
oligo-nucleotide array-comparative genomic hybridization [array-CGH],
the SMC was confirmed to be 18p.
Key Words: Array-CGH, Constitutional disorders, Supernumerary
marker chromosome, Microcephaly, Trisomy 18p.
|
|
S
upernumerary marker chromosomes
(SMC) form a heterogeneous group of chromosomes with an incidence of
0.044% in the general population and increases up to 7 times more frequent
in mentally challenged population(1). This suggests that SMC influences
the clinical manifestation and need proper characterization. Majority of
the SMCs are derived from acrocentric chromosomes having satellited or
bisatellited constriction and around half are derived from chromosome 15.
Non-acrocentric SMCs are comparatively rare and 30% are coupled with
phenotypic effects(2). Various reports describe origin of SMC to be
derived from all the human chromosomes, they are parentally inherited or
de novo and linked with vast clinical features from normal to
extremely mild to severe(3). Recently it has been recognised that every
SMC, though cytogenetically similar, have unique breakpoints(4). This
could be one of the reasons why it is so difficult to build a correct
phenotype-genotype correlation for each marker.
Case Report
The proband, a 13-month-old girl, was referred for
microcephaly and delayed psychomotor development. She was the first child
born to the non-consanguineous parents by cesarean section (birth weight:
2500g; head circumference: 32 cm). She had cranial microcephaly and
neonatal hypotonia followed by hypertonia, jitteriness, apnea, and
seizures. First sleep electroencephalogram report at 11 months showed
epileptiform abnormality from occipital region. She had delayed
psychomotor development first observed at four months and confirmed at 11
months. At the time of presentation, her height was 61.5 cm, head
circumference 42.5cm and weight 4.2 Kg. She had torticollis with deviation
of head to right side, ocular hypertelorism, pseudosquint eyes,
clinodactyly and left simian crease. At the age of 16 months, she could
sit on her own, stand and walk with support.
Cytogenetic study was carried out using peripheral
blood lymphocyte by GTG-banding according to the standard procedure and 50
G-band metaphases were analyzed. Metaphases revealed female karyotype with
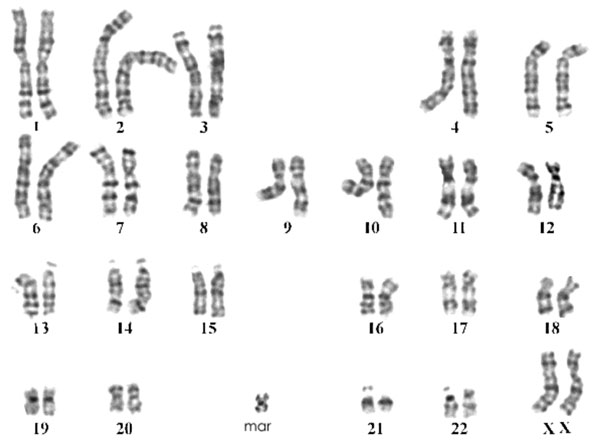
presence of supernumerary marker chromosome in all analyzed cells i.e.
47,XX,+mar (Figure 1). No mosaicism was detected. Parental
karyotype was normal, confirming de novo origin of SMC in the proband. SMC
was further characterized with oligonucleotide array-CGH from Agilent(tm)
using 44,000 probes showing 15 Mb duplication from 18pter to 18p11.21.
However, further confirmation of array-CGH results by FISH was not carried
out.
 |
|
Fig.1 G-banded karyotype showing the marker
chromosome. |
Discussion
SMC occurring as a pure trisomy 18p is an infrequent
observation. The most common phenotypic association is of
non-characteristic facial appearance and subnormal mental development,
foot or hand abnormalities and craniofacial anomalies(5). Till date, 23
cases of trisomy 18p have been published with different origin like
unbalanced segregation due to balanced parental translocation in 10
cases(6), direct tandem duplication in 6 cases(7), complex chromosomal
anomalies in 3 cases(8) and SMC in four cases(5,9,10).
The occurrence of SMC is seven times more in subjects
with developmental delay, which indicates the influence of SMC on
phenotype. Majority of the SMC are acrocentric in origin with 50% from
chromosome 15(9). Some are inherited and transmitted without clinical
manifestations. This is because the gain or loss of p arm of acrocentric
chromosome containing Nucleolar Organizing Region [NOR] region are less
associated with clinically abnormal phenotypic manifestations due to less
functional genes involved in that region [7% vs. 28% derived from non-acrocentric
chromosome](2). In the present case, the SMC is due to trisomy 18p as
revealed by array-CGH study and of de novo origin. The critical
region for most trisomy 18 cases has been demonstrated at 18q while
trisomy of chromosome 18 short arm does not have serious clinical
repercussions. This duplication involves about 67 genes.
The present report demonstrates SMC as trisomy 18p of
de novo origin. It also confirms that trisomy 18p is associated
with subtle phenotypic expression in addition to mild mental retardation.
Moreover, such cases could have been missed in absence of oligonucleotide
micro-array assay.
Acknowledgments
We sincerely acknowledge Manisha Desai and Preeti
Damania for performing cytogenetics analysis. Caroline Thuillier, Alexie
Leurent and Delphine Cerasa for processing the sample for array CGH. Our
thanks to Dr Navin Patel for referring this case.
Contributors: FJS, JA and JJS designed the study
and revised the manuscript for important intellectual content. FJS has
written the manuscript and will act as guarantor.
Funding: Department of Biotechnology [DBT] -
BT/PR9111/MED/12/337/2007, India and UICC, Geneva, Switzerland.
Competing interest: None stated.
References
1. Liehr T, Weise A. Frequency of small supernumerary
marker chromosomes in prenatal, newborn, developmentally retarded and
infertility diagnostics. Int J Mol Med 2007; 19: 719-731.
2. Crolla JA, Youings SA, Ennis S, Jacobs PA.
Supernumerary marker chromosomes in man: parental origin, mosaicism and
maternal age revisited. Eur J Hum Genet 2005; 13: 154-160.
3. http://www.med.uni-jena.de/fish/sSMC/00START.htm.
Accessed January 27th 2009.
4. Kowalczyk M, Srebniak M, Tomaszewska A. Chromosome
abnormalities without phenotypic consequences J Appl Genet 2007; 48:
157-166.
5. Mabboux P, Brisset S, Aboura A, Pineau D, Koubi V,
Joannidis S, et al. Pure and complete trisomy 18p due to a
supernumerary marker chromosome associated with moderate mental
retardation. Am J Med Genet 2007; 143: 727-733.
6. Johansson B, Mertens F, Palm L, Englesson I,
Kristoffersson U. Duplication 18p with mild influence on the phenotype. Am
J Med Genet 1988; 29: 871-874.
7. Li S, Tuck-Muller CM, Martínez JE, Rowley ER, Chen
H, Wertelecki W. Prenatal detection of de novo duplication of the short
arm of chromosome 18 confirmed by fluorescence in situ Hybridization
(FISH). Am J Med Genet 1998; 80: 487-490.
8. Oner G, Jauch A, Eggermann T, Hardwick R, Kirsch S,
Schiebel K, et al. Mosaic rearrangement of chromosome 18:
characterization by FISH mapping and DNA studies shows trisomy 18p and
monosomy 18p both of paternal origin. Am J Med Genet 2000; 92: 101-106.
9. Rodríguez L, Liehr T, Mrasek K, Mansilla E,
Martínez-Fernández ML, Garcia A, et al. Small supernumerary
chromosome marker generating complete and pure trisomy 18p, characterized
by molecular cytogenetic techniques and review. Am J Med Genet 2007;143A:
2727-2732.
10. San Martin V, Fernandez-Novoa C, Hevia A, Novales A, Fornell J,
Galera H. Partial trisomy of chromosome 18(pter—>q11). A discussion on the
identification of the critical segment. Ann Genet 1981; 24: 248-250.
|
|
|
 |
|
