|
|
|
Indian Pediatr 2017;54: 495 -497 |
 |
Kyphoscolitic Type of
Ehlers-Danlos Syndrome with Prenatal Stroke
|
|
Meriem Zahed-Cheikh, *Barthélémy Tosello,
#Stéphanie Coze and
Catherine Gire
From *Department of Neonatologie and #Medical
Imaging Service, Assistance Publique-Hôpitaux de Marseille, Hospital
Nord, 13015 Marseille, France.
Correspondence to: Dr Barthelemy Tosello, Department
of Neonatology, Hospital Nord, Chemin des Bourelly, 13015 Marseille,
France.
Email:
[email protected]
Received: April 15, 2016;
Initial review: August 03, 2016;
Accepted: March 11, 2017.
|
Background: The kyphoscoliotic type of Ehlers-Danlos syndrome (EDS
type VIA) is an autosomal recessive disorder characterized by connective
tissue dysplasia. Case characteristics: We report two children
with perinatal stroke; accompanied by neonatal joint hypermobility,
hypotonia; and early development of kyphoscoliosis. Outcome:
Molecular analysis revealed a PLOD1 gene mutation. Our definitive
diagnosis was a EDS VIA. Message: Prenatal brain stroke is a rare
clinical feature of EDSVIA.
Keywords: Diagnosis, Neonatal hypotonia, PLOD1 gene,
Scoliosis.
|
|
T
he kyphoscoliotic type of Ehlers-Danlos syndrome
(EDS type VIA) is a rare autosomal recessive disorder characterized by
connective tissue dysplasia [1], and is due to a mutation in the
PLOD1 gene [1]. The syndrome’s clinical characteristics are
hypotonia associated with malignant kyphoscoliosis, hyperlaxity,
hyper-elasticity, and skin fragility. Presence of vascular disorder
during the neonatal period does not immediately lend itself to this
diagnosis [2].
We, herein, report two cases of EDS type VIA with
neonatal hypotonia, and prenatal brain stroke.
Case Report
Two siblings born to third degree consanguineous
parents are reported. When Case 1 was 4½ years old, her sister (Case
2) was born. Table I reports the description of the
two cases. An EDS diagnosis was suspected when Case 1 was two
years old. The diagnosis was based on joint hypermobility; delayed motor
development (walking at 24 months, level 2 on Bimanual Fine Function;
and on Gross Motor Function Classification System scales at 24 months).
Suggestive facial dysmorphology, such as blepharochalasis, drooping
cheeks, bluish sclera, tallness with a Marfanoid habitus, and
arachnodactyly also contributed to this diagnosis. Her skin was
hyperelastic with multiple bruises and a lassis venular. Furthermore she
had a ligament laxity at 8/9.
TABLE I Description of Two Cases of Kyphoscolitic Ehlers-Danlos Syndrome
|
Case 1 |
Case
2 |
|
Antenatal data |
Without any complications |
USG: Foot
varus and suspected immobility at 24 wks gestation.` |
|
Mode of delivery |
Spontaneous vaginal delivery |
Induced
vaginal delivery due to immobility and oligohydramnios |
|
Gestational age |
370/6 weeks of gestation |
37 4/6 weeks
of gestation |
|
Apgar score, M1, M5, M10 |
10/10/10
|
10/10/10
|
|
Measures |
Birth weight 2900 g (-1SD), 52 cm
|
Birth weight
298 g (-1SD), 54 cm (+3SD) length, head
|
|
(+2SD) length, head circumference |
circumference of 37 cm (+2SD)
|
|
of 36 cm (+2SD) |
|
|
Physical examination
|
Joint hyperlaxity, hip dislocation, |
Major
generalized neonatal hypotonia, along with
|
|
at birth |
arthrogryposis with talus feet and |
arthrogryposis. Her hands were locked in flexion and
|
|
club hands, and poor gesticulation |
dislocated.
The tegument appearance, joint
|
|
that was contrasted with good visual |
hypomobility,
and dysmorphology all resembled
|
|
contact |
that
of her sister’s
|
|
Brain ultrasonography |
Day 2 of life: IVH Grade IV |
Day 3 of
life: IVH grade IV |
|
Brain MRI (Fig.1) |
Day 3 of life: Parenchymental
|
Day 3 of
life: Parenchymental extension (ischemic
|
|
extension semiovale) of
(the right
|
lesions of
the anterior limb of the left internal capsule
|
|
centrum germinative layer haemorrhage
|
and right
caudate nucleus) of germinative layer haemorrhage
|
|
Evolution
|
5 mo of age: kyphosis, followed by
|
Developed
kyphoscoliosis, required corset at 1 yr age.
|
|
a rapidly progressive kyphoscoliosis |
At age of 5
yr she presented with progressive multiple
|
|
which required a brace, and then a
|
disabilities
such as late walking at 24 months and
|
|
corset, Age 3 yr: surgical
|
diffused
neuromotor disorders based on the
|
|
intervention |
Touwen’s
neurodevelopmental examination. |
|
No anomalies were found on
ophthalmological examination, imaging studies or muscle biopsy.
Karyotype was 46 XX for both. Thrombophilic workup normal. |
After the second child’s birth, our work-up used DNA
blood sampling as well as a skin biopsy taken from the Case 1 to
investigate classic (II) and vascular (IV) types of EDS. The COL3A1,
TGFBR1, and TGBR2 genes exhibited no sequence abnormality.
By using DNA blood samples from sisters, all coding exons and the
neighboring intronic regions of the PLOD1 gene were amplified
from the DNA by PCR (polymerase chain reaction). These were then
sequenced directly with flanking primers and PLOD1 gene dosage
analysis was performed by quantitative Real Time PCR including 19
amplicons in exon 1-19. We then took a blood sample from the parents.
By qPCR a duplication of exons 10 and 16 was detected in the
PLOD1 gene, confirming the diagnosis of EDS type VIA in both
sisters. This duplication was found present on both alleles.
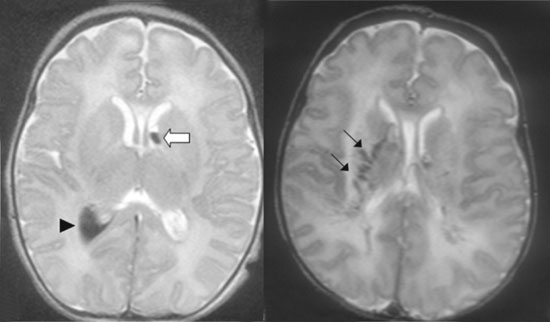 |
|
(a)
(b)
Fig. 1 Brain MRI of cases: (a)
patient 1. Axial T2 sequence showing an cerebral
intraventricular hemorrhage (arrow head) in T2 hypointensity, a
subependymal hemorrhage (white arrow) and hemorrhagic infarction
of the right centrum semiovale with diffusion restriction on
axial DWI MRI images. (b) patient 2. Axial T2 sequences
showing ischemic lesions of the anterior limb of the left
internal capsule and right caudate nucleus, hypoT2 linear images
may result of the periventricular and lenticulostriate vessels
hemorraghic infarction (thin black arrow).
|
|
Discussion
Our report illustrates a rare phenotype of EDS
type VIA with a prenatal brain stroke. When a combination of prenatal
brain stroke and neonatal hypotonia is noted, the possibility of EDS
should always be contemplated. Finding the cause of this hypotonia
requires a rigorous diagnostic approach using the Dubowitz algorithm
[3]. Neonatal hypotonia consistently appeared as a clinical symptom in a
review of 12 cases with variations of kyphoscoliotic EDS phenotype [1].
The presence of prenatal brain stroke and the absence
of kyphoscoliosis noted in the neonatal period evoke an EDS type IV.
This disease, often found in young adults, is linked to mutations in
COL3A1 gene and has a different phenotype [4]. An autosomal
recessive mode of inheritance is most probable, especially with
consanguineous parents, and we carried out COL3A1 genetic
screening by molecular analysis of skin biopsies. These proved to be
negative, and we therefore discarded a diagnosis of EDS type IV [5].
The key diagnostic criteria were severe hypotonia,
tendon laxity, scoliosis and scleral fragility. Assaying the enzymatic
activity of PLOD from a skin biopsy showed 10-16 exon gene
duplication. Neither strokes nor hip dislocation are typical of this
syndrome with hip dislocation being found in only 25% of EDS type VI
cases [1]. Similarly, in EDS type VI there is a possibility of vascular
rupture. A prenatal case is described in a recent review of 15 patients
and in an index case reported by Yis, et al. [6]. Tosun, et al.
[2] suggest that although one of their patient’s subdural and
intraparenchymal hemorrhage could be attributed to a breech delivery or
difficult birth, the patient’s abnormal collagen structure may be a
facilitating factor. Finally vascular ruptures are probably
underestimated in this syndrome. In particular, 15% of mutant mouse
PLOD -/- die of aortic dissections due to smooth muscle and collagen
degeneration in the vessel wall [7]. Thus, a prenatal vascular event
without any etiology with hypotonia and kyphosis ought to prompt a
search for EDS type VI besides, COL4A1/A2 mutations without EDS
were already evaluated in the etiology of intraventricular hemorrhage
detected in utero [4].
Contributors: MZC: participated in the
interpretation and writing of the manuscript; BT, CG, SC: participated
in the patient management, and interpretation and writing of the
manuscript. All the authors approved the final manuscript.
Funding: None; Competing interest: None
stated.
References
1. Rohrbach M, Vandersteen A, Yiº U, Serdaroglu G,
Ataman E, Chopra M, et al. Phenotypic variability of the
kyphoscoliotic type of Ehlers-Danlos syndrome (EDS VIA): clinical,
molecular and biochemical delineation. Orphanet J Rare Dis.
2011;23;6-46.
2. Tosun A, Kurtgoz S, Dursun S, Bozkurt G. A case of
Ehlers-Danlos Syndrome Type VIA with a novel PLOD1 gene mutation.
Pediat Neurol. 2014;51:566-9.
3. Dubowitz L, Ricciw D, Mercuri E. The Dubowitz
neurological examination of the full-term newborn. Ment Retard Dev
Disabil Res Rev. 2005;11:52-60.
4. Parapia LA, Jackson C. Ehlers-Danlos syndrome : A
historical review. Br J Haematol. 2008;141:32-5.
5. Kutuk MS, Balta B, Kodera H, Matsumoto N, Saitsu
H, Doganay S, et al. Is there relation between COL4A1/A2
mutations and antenatally detected fetal intraventricular hemorrhage?
Childs Nerv Syst. 2014;30:419-24.
6. Yiº U, Dirik E, Chambaz C, Steinmann B, Giunta C.
Differential diagnosis of muscular hypotonia in infants: The
kyphoscoliotic type of Ehlers–Danlos syndrome (EDS VI). Neuromus Disord.
2008;18:210-4.
7. Takaluoma K, Lantto J, Myllyharju J. Lysyl
hydroxylase 2 is a specific telopeptide hydroxylase, while all three
isoenzymes hydroxylate collagenous sequences. Matrix
Biology.2007;26:396-403.
|
|
|
 |
|
