|
|
|
Indian Pediatr 2012;49: 490-492
|
 |
Infant with Type A Niemann Pick Disease and
Undetectable Niemann Pick Cells in Bone Marrow
|
|
Sharmila Banerjee Mukherjee, Meenu Pandey, *Seema Kapoor and **T Padma
Priya
From the Department of Pediatrics, Lady Hardinge
Medical College and associated Kalawati Saran Children Hospital, New
Delhi; *Department of Pediatrics, Maulana Azad Medical College and
associated Lok Nayak Hospital, New Delhi; and
**Diagnostics Division, Centre for DNA Fingerprinting and Diagnostics,
Hyderabad, India.
Correspondence to: Dr Sharmila B Mukherjee, Associate
Professor, Department of Pediatrics,
Lady Hardinge Medical College, New Delhi.
Email: [email protected]
Received: August 24, 2011;
Initial review: September 13, 2011;
Accepted: October 19, 2011.
|
Bone marrow aspiration is the preliminary investigation in Niemann Pick
disease type A when enzyme assays and mutation studies are unavailable.
We report an infant with typical phenotype and enzyme deficiency, but
undetectable Niemann Pick cells in the bone marrow. A new mutation R542X
in SMPD gene was also detected.
Key words: Bone marrow, Diagnosis, Niemann pick disease type
A, Storage cells.
|
|
Niemann Pick Disease (NPD)
is a lysosomal storage disorder caused by absence or deficiency of
Acid Sphingomyelinase (ASM), leading to pathological accumulation of
sphingomyelin and cholesterol in the monocyte-macrophage system.
This is characterized by large lipid laden macrophages or Niemann
Pick cells (NP cells) in various tissues. According to clinical
presentation NPD is phenotypically classified as Type A (Classical
infantile neuronopathic form), Type B (Non-neuronopathic visceral
form) and Type C (Juvenile form). We report an infant with NPA, who
despite having typical phenotype and enzyme deficiency, failed to
display NP cells in the bone marrow.
Case Report
A six month old boy presented with gradually
progressive abdominal distension since late neonatal period. There
was no history of persistent fever, vomiting, abnormal bowel
movements, pallor, jaundice, bleeding, rash or additional swelling.
Acquisition of all developmental milestones was delayed. Seizures
and altered consciousness were absent. Antenatal and perinatal
periods were normal. Birth was at term with a weight of 2.3 kg. He
was the second issue of non-consanguineous, healthy, hindu parents.
A male sibling had expired at 14 months of age with similar illness.
Anthropometry was within normal range for age,
with weight 6.6 Kg (83.5 % of 50th percentile of WHO child growth
standards), length 65.4 cm (96.75% of 50 th
percentile of WHO child growth standards) and head circumference
42.5 cm (between 10th
and 25th percentile).
The facies appeared coarse with a broad forehead, depressed nasal
bridge, thick lips and anteverted nostrils. There was no icterus or
lymphadenopathy. The abdomen was protuberant with firm
hepatosplenomegaly (liver span was 11 cm in mid clavicular line and
spleen size was 6 cm in splenic axis below costal margin) and no
free fluid. Salient neurological findings were a conscious but
apathetic infant with cherry red spots, normal cranial nerves and
power, generalized hypotonia and hyporeflexia with extensor plantar
responses. Structured developmental assessment demonstrated a
Development Quotient of 62, suggestive of mild global developmental
delay. Differential diagnoses of Niemann-Pick disease, Sandhoff
disease and GM1 gangliosidosis were considered in order of
suspicion.
Investigations revealed mild normocytic
normochromic anemia (Hb 9.5 gm/dL) with normal total and
differential leukocyte, platelet and reticulocyte counts. Liver
function tests were deranged; total bilirubin 1.4 mg/dL, direct
bilirubin 0.4 mg/dL, Aspartate amniotransferase 790 IU. Alanine
amniotransferase 907 IU, alkaline phosphatase 1481 IU). Serum
cholesterol level was 180 mg/dl (normal for age 65-175 mg/dL).
Abdominal ultrasonography confirmed liver and spleen enlargement
with normal echotextures and normal portal vein diameter. Hearing
and visual evaluation (by BERAphone screening and Visual Evoked
Response) and skeletal survey were normal. Bone marrow aspiration
revealed cellular bone marrow with normoblastic erythroid series,
myeloid series and megakaryocytes. Storage cells were not detected
even on bone marrow biopsy. Thyroid Function tests and MRI cranium
were normal. Sequential enzyme assays were planned. Serum levels of
ASM were undetectable which was diagnostic of Niemann-Pick disease,
the phenotypes suggestive of type A (Classical infantile
neuronapathic form).
During follow up, neuro-developmental status
remained static. Hepatosplenomegaly progressively increased but
without further enzyme derangement. At 11 months, he contracted
severe pneumonia and succumbed enroute to hospital. Permission could
be obtained only for a post mortem liver biopsy, the histopathology
of which revealed NP cells. Gene sequencing of the Spingomyelin
phosphodiesterase 1 (SMPD1) gene revealed compound
heterozygosity for stop codon mutations R443X and R542X in exon 3
and 5, respectively (Fig. 1). Origin was maternal in
the former and paternal in the latter.
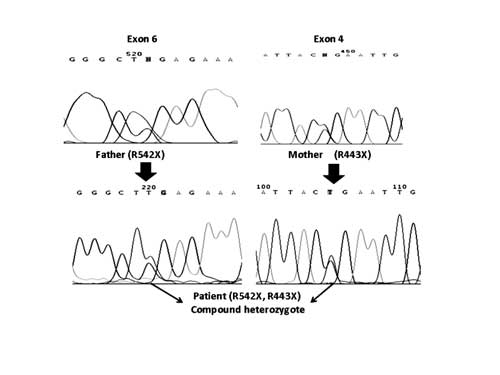 |
|
Fig. 1 Gene sequencing of patient
demonstrating paternal origin of mutation R542X and maternal
origin of R443X.
|
Discussion
In type A and B NPD, the affected enzyme is
encoded by the SMPD1 gene located on chromosome bands
11p15.1-p15.4, resulting in primary ASM deficiency with activity
1-10% of normal. Pathological sphingomyelin deposition results in
infiltration of bone marrow, spleen, liver and lymph nodes with NP
cells. In developed countries, diagnosis is established by enzyme
assay and mutation analysis, rather than more invasive alternatives
like BMA. However, in developing countries, these modalities are
expensive and not easily available. Common practice in such
circumstances is demonstration of NP cells by BMA.
Organomegaly has been reported as the commonest
presentation of NPD-A, with a median age of diagnosis 6 months [1].
Elevated cholesterol was considered an early marker of the disease
[1]. Since reticuloendothelial organs are completely infiltrated
with NP cells originating from lipid-accumulating bone marrow
progenitor cells, non-demonstration of NP cells in symptomatic
patients is unusual. Extensive literature revealed no prior studies
reporting absence of NP cells in BMA. Suboptimal sensitivity of BMA
has been previously reported in children with type C NPD presenting
with cholestatic jaundice. The overall sensitivity of 64% decreased
to 57% when BMA was performed during the first year [2]. This can be
explained by the later age of onset in type C. The clinical profile
of our case (static neuro-developmental status, hepatosplenomegaly
without increasing enzyme deterioration and normal cholesterol)
reflects early illness with probably less extensive infiltration of
the bone marrow. This questions the reliability of BMA in early
illness or young infants with NPD-A.
Mutational analysis has revealed many disease
associated alleles in NPD-A. Most are sporadic with only a few
having ethnic predilection like p.R496L, p.L302P and p.P330fs in
Ashkenazi population and c.677delT in Israeli Arabs [3]. Most
mutations are single base substitutions and small deletions with or
without a frameshift [4]. Small deletions or nonsense mutations
result in truncated ASM polypeptide and missense mutations render
the enzyme non-catalytic in NPD-A, whereas enzyme is defective with
residual catalytic activity and milder phenotype in NPD-B [5]. In
this case, mutation R542X (arginine to stop codon at amino
acid 542) is novel whereas mutation R443X (arginine to stop codon at
amino acid 443) has been reported earlier in a homoallelic patient,
also of Indian origin [6]. Only further studies will be able to
confirm a possible Indian predilection.
In resource limited settings, demonstration of
storage cells on BMA is the preliminary investigation despite its
invasiveness. Confirmatory enzyme assays are performed subsequently.
Absence of NP cells in the bone marrow usually leads to considering
alternative diagnoses. When strong clinical suspicion of NPD-A
exists, a normal BMA should not exclude the diagnosis without an
enzyme assay. If services are unavailable locally, blood can be
collected as ‘spots’ on 903S&S filter paper (GE) which remain
sufficiently stable to be transported to the testing laboratory for
enzyme analysis [7].
Acknowledgements: Dr Ashwin Dalal,
Director, Diagnostics Division, Centre for DNA Fingerprinting and
Diagnostics, Hyderabad for providing his valuable expertise.
Contributors: SBM, MP and SK were involved in
clinical diagnosis and manuscript writing. SBM and SK will stand as
guarantors. TPP was involved in the genetic tests and their
interpretation. All authors approved the final manuscript.
Funding: None; Competing interests:
None stated.
References
1. McGovern MM, Aron A, Brodie SE, Desnick RJ,
Wasserstein MP. Natural history of Type A Niemann-Pick disease:
Possible endpoints for therapeutic trials. Neurology. 2006;
66:228-32.
2. Rodrigues AF, Gray RG, Preece MA, Brown R,
Hill FG, Baumann U, et al. The usefulness of bone marrow
aspiration in the diagnosis of Niemann-Pick disease type C in
infantile liver disease. Arch Dis Child. 2006;91:841-4.
3. Ricci V, Stroppiano M, Corsolini F, Di Rocco
M, Parenti G, Regis S, et al. Screening of 25 Italian
patients with Niemann Pick A reveals fourteen new mutations, one
common and three private in SMPD1. Hum Mutat.2004;24:105.
4. Sikora J, Pavlu-Pereira H, Elleder M, Roelof
H, Wever RA. Seven novel acid sphingomyelinase gene mutations in
Niemann Pick type A and B patients. Ann Hum Genet. 2003;67:63-70.
5. Takahashi T, Suchi M, Desnick RJ, Takada G,
Schuchman EH. Identification and expression of five mutations in the
human acid sphingomyelinase gene causing types A and B Niemann-Pick
disease: molecular evidence for genetic heterogeneity in the
neuronopathic and non-neuronopathic forms. J. Biol. Chem.
1992;267:12552-8.
6. Schuchman E. Two new mutations in the Acid
sphingomyelinase gene causing type A niemann-Pick Disease: N389T and
R441X. Hum Mutat. 1995;6:352-54.
7. Chamoles NA, Blanco M, Gaggioli D, Casentini C.
Gaucher and Niemann-Pick diseases—enzymatic diagnosis in dried blood
spots on filter paper: retrospective diagnoses in newborn-screening
cards. Clin Chim Acta. 2002;317:191-7.
|
|
|
 |
|
