b-thalassemia
is a monogenic disorder characterized by reduced or absent
synthesis of the b-globin
chain, one of the main components of adult hemoglobin (HbA,
a2b2).
Several hundred mutations (both point mutations and deletions)
are now described in the human
b-globin gene
(HBB gene cluster on chromosome 11, Fig. 1) or its
regulatory elements, leading to decreased (b+
genotype) or absent (b0
genotype) synthesis of the b-globin
[1]. This results in a relative increase in the unattached
a
chains (a/b-chain
imbalance) that form insoluble hemi-chromes in the erythrocyte
progenitors. The hemi-chromes damage the erythrocyte membrane,
leading to severe intramedullary erythrocyte apoptosis
(ineffective erythropoiesis) and severely shortened red blood
cell (RBC) life span due to extra-medullary hemolysis, leading
to severe anemia (low hemoglobin, Hb) [2,3].
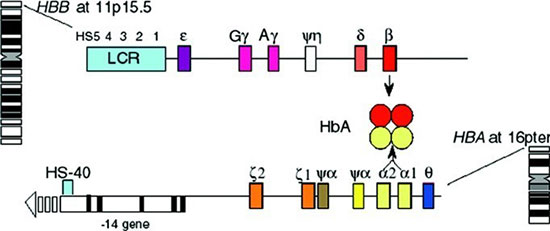 |
|
Fig. 1 HBB gene cluster
(b-globin gene) and HBA gene cluster (a-globin
gene); transcribed globin proteins combine to make adult
hemoglobin (HbA, µ2b2).
|
The phenotype of
b-thalassemia
is variable depending upon the reduction (a+/b+)
or complete absence of b-globin
chain synthesis (b0/b0)
and other genetic variables like co-inheritance of
a- and
g-mutations,
as well as co-inheritance of other hemoglobinopathies (e.g. HbE,
Lepore and sickle hemoglobin) [4,5]. Some mutations also alter
the fetal hemoglobin (HbF, a2g2)
to HbA switch and may lead to higher production of HbF into
adulthood (hereditary persistence of fetal Hb, HPFH) resulting
in less severe anemia [6,7]. Therefore, though the severity of
thalassemia can be usually predicted based on the mutation
analysis of the HBB cluster, other genetic factors may
modify the actual phenotype and transfusion requirements.
Although the switch from
g-
to
b-globin
synthesis begins before birth, complete replacement of the HbF
by HbA occurs in the postnatal period. Consequently, infants
with severe b-globin
chain abnormality become trans-fusion-dependent around 6 months
of age, when levels of HbF decrease significantly. Based on
their transfusion needs, b-thalassemia
patients are classified as trans-fusion-dependent thalassemia
(TDT) or non-transfusion-dependent thalassemia (NTDT), although
these defini-tions are also fluid, as some NTDT patients may
need regular transfusions as they become older [8].
b
-thalassemia has a
high global incidence, especially in Asia (northern and eastern
India) and Eastern Mediter-ranean regions. The conventional
management of patients affected by the severe form of the
disease relies on chronic and regular blood transfusions (every
3-4 weeks) to maintain nadir hemoglobin at or above 9 g/dL along
with iron chelation therapy, to prevent the toxicities of iron
overload [3,9].
Currently, the only curative therapy is
allogeneic hematopoietic stem cell transplant (HSCT) from an
HLA-matched sibling or unrelated donor or cord blood unit
(located through national bone marrow donor registries), with
good outcomes [10]. HSCT is recommended in relatively younger
patients, prior to development of increased iron overload in
organs (especially, liver and myocardium) and when suitable
HLA-matched donors are available to decrease the risks of
toxicity and graft-versus host disease (GVHD). Disease-free
survival exceeds 85%, depending on patients’ age, HLA-matching
and clinical factors like iron overload, liver fibrosis and
hepatomegaly [11,12]. Full matched sibling donor HSCT in younger
children (<16 years of age) is considered standard of care,
while alternative donor HSCT (from mis-matched unrela-ted or
haploidentical donors) are still experimental, and are not
devoid of complications like rejection, viral reacti-vations,
and graft versus host disease (GVHD) [13-15].
Gene Therapy for Thalassemia
The era of genome sequencing, understanding
of the HBB gene cluster and its strict regulation and
control, along with advancements in vector development and
gene-editing platforms, has provided new options for the
treatment for thalassemia patients. The expression of
b-like genes
is regulated by a locus control region (LCR) via
looping-mediated interactions with the globin promoters,
therefore these LCR and promoter regions are essential for the
globin gene expression [16]. The comp-lete understanding of the
switch from g-globin
to b-globin
production during infancy, and the control of this switch by
various transcription factors (TF) has provided new targets for
gene-modifications. Speckle-type POZ protein (SPOP), globin
transcription factor 1 (GATA-1) and B-cell Leukemia/Lymphoma 11A
(BCL11A) are now recognized as important TFs, that bind to
specific sites in the HBB gene and control the switch
from production of HbF to HbA [17-19].
Advances in vector development, transduction
of human stem and progenitor cells (HSPCs) and various
gene-editing tools, provide a new hope for availability of
curative options soon, making gene-therapy one of the most
promising treatment options.
The goal of current gene therapy strategies
is to induce production of b-
or g-globin,
thereby decreasing the levels of unattached
a-globin
chains, to restore the alpha/non-alpha globin ratio in RBCs.
This should lead to correction of ineffective erythropoiesis and
improved RBC lifespan (decreased hemolysis), with larger number
of erythrocytes with higher hemoglobin surviving longer in the
peripheral blood, leading to the correction of anemia and
reduction in transfusion needs [20-23].
The common treatment schema of patients
under-going autologous gene modification and infusion is shown
in Fig. 2. Major steps in the treatment are explained
below.
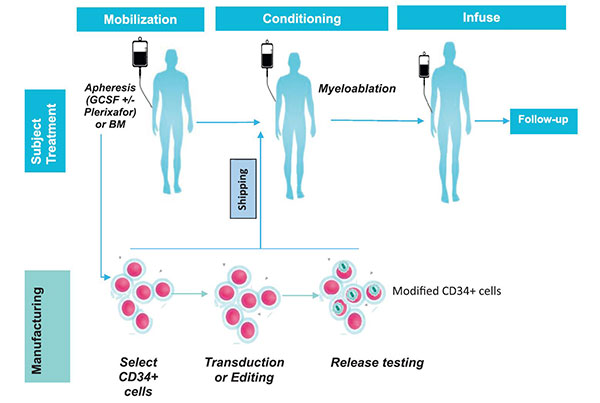 |
|
Fig. 2 Overview of treatment
plan for gene-modified human stem and progenitor cells.
|
Stem cell mobilization and collection:
G-CSF and plerixafor mobilized HSPCs (HPC-Apheresis) are
obtained in TDT patients by apheresis procedure as the starting
material for gene-modification. Plerixafor is added upfront in
the collection protocols, as it leads to efficient mobilization
of large number of stem cells in the periphery and decreases the
number of collection days and procedures needed for adequate
number of stem cells to be collected for gene modifications
[24,25]. Adequate HSPCs can be collected from the TDT patients
using this combination, despite G-CSF dose reductions
recommen-ded in post-splenectomy patients to avoid
hyper-leukocytosis. HSPCs are collected via the leukapheresis
procedure and large volumes of blood (~15-20 L of recirculated
blood volume for adults or approximately four total blood
volumes in younger patients) should be cycled per day, based on
patient tolerability. Average HPC-A collections over 2-3 days in
thalassemia patients can yield approximately 15-50×106
CD34+/kg (based on experience from early clinical trials) which
are adequate for manufacturing and for storing a small fraction
(³2
×106 CD34+/kg cells)
as an unmanipulated ‘back-up’, as a safety precaution in case of
non-engraftment with modified HSPCs. The collected HSPCs undergo
CD34+ enrichment process, prior to undergoing
gene-modi-fication.
Myeloablation: Efficient myeloablation of
the expanded erythroid pool in the bone marrow of the TDT
patients is essential to create adequate space in the bone
marrow niches for adequate engraftment of gene-modified HSPCs,
as the gene-modified HSPC do not have a selective survival
advantage in thalassemia over the non-gene-modified HSPC.
Busulfan is currently the best agent to achieve myeloablation,
as the dose can be tailored for each patient based on first dose
pharmacokinetics to achieve a standardized target dose range
required for myeloablation and to avoid excessive
extra-medullary toxicity and lymphodepletion.
Infusion of gene-modified stem cells: The
gene-modified HSPCs are usually cryopreserved in 5% dimethyl
sulfoxide (DMSO) solution. Once the final product meets all the
release criteria (sterility, viability, purity, and %
gene-editing frequency or vector copy number [VCN] for
gene-insertions), and minimum cell dose criteria (> 2-3× 106
CD34+/kg) needed for hematopoietic engraftment, the
cryopreserved cells are transported to the treatment site. The
cells are thawed and infused intravenously as per standard
infusion procedure for autologous stem cell transplants.
Post-transplant care: Care in a
specialized BMT unit is recommended, as these patients do become
neutropenic and need transfusion support (packed red blood cells
and platelet transfusions). Close monitoring and supportive care
for busulfan related side-effects, especially mucositis, nausea,
infections and veno-occlussive disease (VOD) of liver is
recommended. Patients are discharged once they achieve
neutrophil engraftment, can eat, drink, and retain their
prophylactic medications. Since busulfan is myelo-ablative but
does not cause severe lymphopenia, infection prophylaxis is only
recommended for a short period post-transplant. Currently, a 15
years follow up is required for all gene-therapy trials as
dictated by regulatory agencies in US and EU. This long follow
up is required to ascertain the durability and safety of these
experimental approaches.
Currently, the gene-therapy approaches can be
divided into two broad groups viz., gene-insertion, and
gene-editing approaches.
Gene-Insertion
This involves insertion of a lentiviral or
retroviral vector, that contains the whole regulatory machinery
and the
b-or
g-globin
producing genes, into autologous HSPCs ‘ex-vivo’, and then
infusing these modified HSPCs back to the patient after
myeloablation [26-28]. Though conceptually straightforward, the
field has techno-logically advanced only recently, where the
vectors (packaged with the large HBB gene and its
regulatory elements- promoter, enhancer and parts of LCR) can
now be produced at a large scale, achieve high levels of
purification and potency to transfect large number of
‘non-proliferating’ human stem cells to provide clinical
meaningful responses [22,29]. For a long-lasting correction and
life-long production of erythrocytes (with the hope of one-time
curative treatment), the insertions are done in HSPCs (CD34+
enriched popu-lation, Milteyni), which includes the long-term
repo-pulating subsets of stem cells. For gene insertion into
stem cells, the globin producing genes are placed under the
control of an erythroid-specific promoter, so that the
transcription of the inserted genes can only occur in erythroid
precursors, and not in white blood cells or platelets, which are
also derived from the modified hematopoietic stem cells [30].
There are multiple designed lentiviral
vectors in clinical trials now for
b-thalassemia
(Web Table I). Once a significant number of HSPCs have
been transduced and infused back to a patient, it is expected
that the erythrocyte progenitors derived from these modified
stem cells will produce enough
b- (or
g) globin
(depending on the insertion) to combine with
a-chains and
reduce the a/b
imbalance.
Risks of Gene-Insertion
Since the vector insertions into the stem
cells occur randomly and remains largely an uncontrolled
process, there is a small risk that some insertions into human
stem cells can occur near proto-oncogenes and can stimulate
clonal proliferation leading to leukemia/myelodysplastic
syndrome (MDS) [31-33]. With the new optimized and
self-inactivating (SIN) lentiviral vectors, the insertions into
the human stem cells occur ‘semi-randomly’ i.e. lentivirus
insertions occur at preferential sites in the transcription
units of human genome, but still lead to polyclonal
reconstitution, compared to retroviral vectors that were
associated with high risk of insertional mut-agenesis [34]. All
clinical trials currently perform integration site analysis to
monitor patients of any emerging clonal population. Currently,
regulatory agencies require all patients treated with gene
therapies to be followed for a total period of 15 years, to
clearly establish the incidence of this risk. Fortunately, till
date, none of the patients treated with lentiviral vectors have
developed any leukemia or MDS related to lentiviral vector
insertions [35].
Results of gene-insertion clinical trials:
All patients treated recently have tolerated the
conditioning regimen with myeloablative doses of busulfan
without any unexpected toxicity. Approximately 10% of patients
are reported to have developed mild to moderate veno-occlusive
disease (VOD) of the liver related to underlying liver fibrosis
but have responded to supportive care or defibrotide treatment.
In the early Phase 1/2 trials, all patients had engrafted,
though efficacy analysis of the first few patients treated with
BB305 lentiviral vector, showed variable responses and total
hemoglobin production. This variability is expected, as patients
with
b-thalassemia
have large genetic heterogeneity due to varied mutations in the
HBB cluster and various genetic modifiers and therefore,
the level of hemoglobin required to become transfusion
independent is variable. The initial results of two concurrent
trials (HGB 204 and 205 using BB305 vector), show an average
production of 4-5 g/dL of HbAT87Q
from the gene-insertions (HbAT87Q
is the gene-insertion derived HbA that can be detected
separately from transfusion derived HbA by HPLC due to presence
of one amino-acid substitution: Threonine at 87 position instead
of Glutamine) [30]. An increase of hemoglobin by ~5 g/dL is
enough to lead to transfusion independence in HbE/b-thalassemia
and b0/b+
patients, but only leads to decrease in transfusion requirements
in b0/b0
patients, where there is a need for higher levels of hemoglobin
production to become transfusion independent [22]. Ninety
percent (18/20 with >3 months follow up) of non-b0/b0
patients treated show rapid rise in gene-derived hemoglobin (HbAT87Q)
production post-treatment, maintaining total hemoglobin levels
of >9 g/dL (mean 11.6 g/dL; range 9.3-13.3g/dL), with
transfusion independence [36]. Based on early encouraging
results and safety profile, the lentiglobin gene therapy
(Zynteglo) was conditionally approved in EU in June, 2019 for
TDT patients with non-b0/b0
genotype who are ³12
years of age (this is still not approved by FDA in US). The
results for the b0/b0
patients are still under study (HGB 212 trial, NCT 03207009),
but do show variable results with 8/11 patients followed for >3
months maintaining hemoglobin above 9 g/dL, though it is still
early to comment on durability of the outcomes at this stage
[37].
Gene-Editing
Availability of new tools and techniques in
the last few years is leading to a rapid development of
gene-editing approaches to ameliorate the anemia in thalassemia
patients. Last few years have seen advances in availability of
different engineered nucleases – zinc-finger nucleases (ZFN),
transcription activator-like effector nucleases (talens), and
clustered regularly interspaced short palindromic repeats
(Crispr)-associated-nuclease 9 (Crispr- Cas9), which are
nucleases that act like mole-cular scissors and cut the human
DNA at precise locations [38-40]. These nucleases differ in
their precision, specificity, efficiency, and ability to make
single versus double stranded edits in the target sequence of
DNA. The major differences between gene-insertion and gene-
editing platforms are highlighted in Table I.
 |
Of these techniques, Crispr-Cas9 is the most
appealing, as it leads to precise double stranded breaks in the
DNA helix, using a pre-designed 42-nucleotide guide sequence
(Crispr guide), which has bases complimentary to the target site
of the desired break in the DNA [41-43]. The guide carries the
Cas-9 nuclease to the target location in the genome to make
small edits. Electroporation of Cas9 nuclease and single guide
RNA (sgRNA) as a ribonucleo-protein (RNP) complex leads to
efficient delivery of genome editing material into HSPCs [44].
BCL11A (the TF that controls the switch from
HbF to HbA and functions as a repressor of HbF) provides an
excellent target for gene-editing approaches for
hemo-globinopathies [45,46]. By suppressing BCL11A TF, it is
postulated that HbF production can be triggered again in
thalassemia patients to a sufficient degree to ameliorate anemia
and avoid transfusions.
Making specific deletions in the erythroid
specific enhancer region of the BCL11A gene is a
promising approach that is being explored currently [47,48]. Two
programs to treat TDT are using either ZFN or Crispr-Cas9
platforms to make small deletions in the erythroid specific
enhancer region of the BCL11A gene located on Chromosome
2. The major advantage of these platforms is that they do not
directly make edits in the HBB gene, as they target the
BCL11A gene, allowing the endogenous regulation and
sustained production of the globin proteins to continue. These
clinical trials are currently recruiting patients.
Another approach to increase HbF production
is to recreate the mutations seen in patients with HPFH by
making gene edits in the HBB gene. This is achieved by:
i) creating small deletions e.g. in the
g-d
intergenic region leads to significant enhancement of the
g-gene
expression [6]; ii) creating small deletions in the area
of HBB cluster where BCL11A binds (e.g. CCAAT box
region), so the effect of TF can be inhibited [49,50]; and
iii) creating point mutations in the
b-globin
promoter region that can also lead to over expression of the
mutated gene [51].
Pre-clinical studies are currently ongoing
using Crispr-Cas12 platform to perform edits in the CCAAT box of
the HBB gene, which overlaps with the BCL11A TF binding
site, to increase the levels of HbF. This approach requires a
higher degree of precision (‘on-target’ activity), so as not to
disrupt the endogenous production of globin proteins. Both the
gene-insertion and gene-editing methods, now scaled to human
applications, are in multiple clinical trials now (Web Table
I).
Pros and Cons of Gene-Editing Strategy
The main advantage of gene-editing
(especially Crispr-Cas9 or Cas12) platform is the high
efficiency and precision of the gene-edits made in the defined
DNA locus [52].The main drawback of gene-editing nucleases is
that they can make unintended edits in other parts of the
genome, what is called ‘off-target’ activity [53,54]. Despite
their design for accurate target gene editing, unintended
off-target interactions between nucleases and genome sequences
can still occur. There are multiple cell based and in-vitro
assays and computational strategies designed to assess the
off-target activity of the guides and nucleases and to predict
their functional importance during pre-clinical assessments
[55-58]. The goal of these pre-clinical assessments is to define
the efficiency of ‘on-target’ editing and ascertain risks of
‘off-target’ activity (if any) of a Crispr guide.
In addition to potential off-target activity,
chromosomal rearrangement events can also occur, due to double
stranded breaks induced during gene-editing [59]. There-fore,
serial karyotype analysis is also important during follow-up to
analyze chromosome instability of gene-editing platforms.
The assessment of on-target, off-target and
geno-toxicity assays done in the gene-editing platforms is
specific to the guide and the nuclease used to make the gene
edits. Unlike the gene-insertion trials using lentiviral
vectors, the safety profile of the gene-editing techniques
cannot be generalized, as it is specific for the guide and
nuclease. Therefore, it is essential to keep this caveat in mind
when comparing adverse events of one gene-editing clinical trial
with another.
Long-term assessments of safety in clinical
trials is still the gold standard compared to the computational
models for analyzing off-target activity currently available
[55-57], as detection of an ‘off-target’ site activity for a
guide does not necessarily mean it will lead to a clinically
meaningful adverse event.
Results of gene-editing clinical trials:
Gene editing is currently undergoing phase I trials in humans.
The results of the first patient (
b0/IVS-1-110
genotype) treated with Crispr-Cas9 gene editing (CTX001 product)
at 12-months post-treatment show that the patient is transfusion
independent, with total hemoglobin level of 12.7 g/dL (12.4 g/dL
of Hb-F), and 99% erythrocytes in peripheral blood expressing
high levels of HbF (F cells) [60].
Therefore, it is essential to recognize that
long-term safety, durability, with continued transfusion
independence and improvement in quality of life with no further
requirement for chelation therapy, will decide which of these
platforms lead to optimal risk-benefit ratio for acceptability.
PEDIATRIC PERSPECTIVES
Most of the Phase I human clinical trials of
gene therapy are initiated first in adult patients (>18 years of
age) who can understand the risks and benefits of these
approaches clearly and consent to the experimental treatment.
Once safety is established in the initial cohort of adult
patients, the age can be lowered to include younger patients.
Currently, lentiviral gene insertion trials are enrolling
patients >12 years of age and the goal is to follow similar
regulatory strategy for other gene-editing trials, once the
initial safety data is available. The younger age limit needs to
be established, as it is not the busulfan toxicity, but the
risks of HSPCs collection via apheresis procedures in very young
patients (currently safety is established for patients >20 kg
without requiring any blood priming or other safety
precautions).
Since the ‘off-target’ effects of many of
these gene-editing strategies and the risk of insertional
oncogenesis may require a longer duration of follow-up in
pediatric patients to establish safety, therefore it is expected
that for many of these new experimental trials it may take
longer time for safety to be established prior to approval in
younger patients.
It is also expected that younger patients may
tolerate busulfan myeloablation much better than older patients
with organ dysfunctions related to iron overload, although the
issue of fertility cryopreservation needs to be discussed with
individual families as part of the consent process (as
infertility is a common long-term toxicity of busulfan and sperm
or egg cryopreservation options may be limited in younger
patients compared to adults). It is to be noted that the risks
of infertility also exist with allogeneic HSCT where
chemotherapy based conditioning regimens are utilized.
It is also important to note that correction
of ineffective erythropoiesis is an important treatment goal
for young patients, other than transfusion independence, to
avoid complications of NTDT later in life.
Hence, it is envisioned that gene therapy may
provide an alternative option of treatment for younger patients
with TDT, especially in patients who lack well matched (HLA)
family donors and in countries where large natio-nal bone marrow
donor registries or cord blood banks do not exist.
CONCLUSIONS
Recent advances in whole genome sequencing,
an understanding of the control and regulation of HBB
gene along with improvements in vector biology and
manufacturing, availability of new gene-editing nucleases that
can lead to sufficient degree of gene modifications in HSCs to
achieve meaningful clinical benefit, has recently led to
multiple active clinical trials in patients. The early data from
these experimental trials looks promising with potential to lead
to a long-term durable transfusion independence and one therapy
has already been approved in EU for TDT patients >12 years of
age for non-
b0/b0
patients. There is a hope that with the continued analysis of
safety, durability and with continued refinement of
manufacturing with improved efficiencies, gene therapies could
potentially address the global health burden of
b-thalassemia.
Note: Supplementary material related to
this study is available with the online version at
www.indianpediatrics.net
Competing interests: The author is also
employed by Crispr Therapeutics Inc. that sponsors the CTX001
thalassemia trial. Only publicly available information has been
provided and the manuscript was not influenced in any way by
this relationship. Part of the text in this manuscript was
adapted for pediatrics audience from previously submitted
reviews to other journals by the author.
Funding: None.
REFERENCES
1. Giardine B, Borg J, Viennas E, et al.
Updates of the HbVar database of human hemoglobin variants
and thalassemia mutations. Nucleic Acids Res.
2014;42:D1063-9.
2. Rund D, Rachmilewitz E.
Beta-thalassemia. N Engl J Med. 2005;353:1135-46.
3. Rachmilewitz EA, Giardina PJ. How I
treat thalassemia. Blood. 2011;118: 3479-88.
4. Thein SL. Genetic modifiers of
beta-thalassemia. Haemato-logica. 2005;90:649-60.
5. Danjou F, Anni F, Galanello R.
Beta-thalassemia: From genotype to phenotype. Haematologica.
2011;96:1573-5.
6. Bernards R, Flavell RA. Physical
mapping of the globin gene deletion in hereditary
persistence of foetal haemoglobin (HPFH). Nucleic Acids Res
1980;8:1521-34.
7. Forget BG. Molecular basis of
hereditary persistence of fetal hemoglobin. Ann NY Acad Sci.
1998;850:38-44.
8. Musallam KM, Rivella S, Vichinsky
Rachmilewitz EA. Non-transfusion-dependent thalassemias.
Haematologica. 2013; 98: 833-44.
9. Cao A, Galanello R. Beta-thalassemia.
Genet Med. 2010; 12: 61-76.
10. Angelucci E, Matthes-Martin S,
Baronciani D, et al. Hematopoietic stem cell
transplantation in thalassemia major and sickle cell
disease: indications and management recommendations from an
international expert panel. Haemato-logica, 2014;99: 811-20.
11. Lucarelli G, Isgrò A, Sodani P,
Gaziev J. Hematopoietic stem cell transplantation in
thalassemia and sickle cell anemia. Cold Spring Harb
Perspect Med. 2012;2: a011825.
12. Locatelli F, Kabbara N, Ruggeri A,
et al. Outcome of patients with hemoglobinopathies given
either cord blood or bone marrow transplantation from an
HLA-identical sibling. Blood. 2013; 122:1072-8.
13. Sodani P, Isgrò A, Gaziev J, et al.
Purified T-depleted, CD34+ peripheral blood and bone marrow
cell transplantation from haploidentical mother to child
with thalassemia. Blood, 2010;115: 1296-302.
14. Bertaina A, Merli P, Rutella S, et
al. HLA-haploidentical stem cell transplantation after
removal of alphabeta+ T and B cells in children with
nonmalignant disorders. Blood. 2014;124: 822-6.
15. Soni S, Breslin N, Cheerva A.
Successful unrelated umbilical cord blood transplantation
for class 3 beta-thalassemia major using a reduced-toxicity
regimen. Pediatr Transplant. 2014; 18:E41-3.
16. Kim A, Dean A. Chromatin loop
formation in the beta-globin locus and its role in globin
gene transcription. Mol Cells. 2012;34:1-5.
17. Lan X, Khandros E, Huang P, et al.
The E3 ligase adaptor molecule SPOP regulates fetal
hemoglobin levels in adult erythroid cells. Blood Adv.
2019;3:1586-97.
18. Masuda T, Wang X, Maeda M, et al.
Transcription factors LRF and BCL11A independently repress
expression of fetal hemoglobin. Science. 2016;351:285-9.
19. Sankaran VG, Menne TF, Xu J, et al.
Human fetal hemoglobin expression is regulated by the
developmental stage-specific repressor BCL11A. Science,
2008; 322:1839-42.
20. Breda L, Carla Casu, Sara Gardenghi,
et al. Therapeutic hemoglobin levels after gene transfer in
beta-thalassemia mice and in hematopoietic cells of
beta-thalassemia and sickle cells disease patients. PLoS
One, 2012;7: e32345.
21. Bank A, Dorazio R, Leboulch P. A
phase I/II clinical trial of beta-globin gene therapy for
beta-thalassemia. Ann N Y Acad Sci. 2005;1054:308-16.
22. Thompson AA, Walters MC, Kwiatkowski
J, et al. Gene therapy in patients with
transfusion-dependent beta-thalassemia. N Engl J Med.
2018;378:1479-93.
23. Roselli EA, Mezzadra R, Frittoli
MC, et al. Correction of beta-thalassemia major by gene
transfer in haematopoietic progenitors of pediatric
patients. EMBO Mol Med, 2010;2:315-28.
24. Yannaki E, Karponi G, Zervou F, et
al. Hematopoietic stem cell mobilization for gene therapy:
superior mobilization by the combination of
granulocyte-colony stimulating factor plus plerixafor in
patients with beta-thalassemia major. Hum Gene Ther.
2013;24: 852-60.
25. Yannaki E, Papayannopoulou T, Jonlin
E, et al. Hematopoietic stem cell mobilization for gene
therapy of adult patients with severe beta-thalassemia:
results of clinical trials using G-CSF or plerixafor in
splenectomized and nonsplenectomized subjects. Mol Ther.
2012;20:230-8.
26. Cavazzana M, Mavilio F. Gene Therapy
for Hemoglobinopathies. Hum Gene Ther. 2018.
27. Dong A. Rivella S, Breda L. Gene
therapy for hemoglobinopathies: Progress and challenges.
Transl Res. 2013;161: 293-306.
28. Malik P, Arumugam PI. Gene therapy
for beta-thalassemia. Hematology Am Soc Hematol Educ
Program. 2005: p. 45-50.
29. Negre O, Eggimann AV, Beuzard Y, et
al. Gene therapy of the beta-hemoglobinopathies by
lentiviral transfer of the beta (A(T87Q))-globin gene. Hum
Gene Ther. 2016;27:148-65.
30. Payen E, Colomb C, Negre O, Beuzard
Y, Hehir K, Leboulch P. Lentivirus vectors in
beta-thalassemia. Methods Enzymol. 2012; 507:109-24.
31. Ginn SL, Liao SVL, Dane AP, et al.
Lymphomagenesis in SCID-X1 mice following
lentivirus-mediated phenotype correction independent of
insertional mutagenesis and gammac overexpression. Mol Ther.
2010;18:965-76.
32. Hacein-Bey-Abina S, Garrigue A, Wang
GP, et al. Insertional oncogenesis in 4 patients after
retrovirus-mediated gene therapy of SCID-X1. J Clin Invest.
2008; 118:3132-42.
33. Moiani A, Paleari Y, Sartori D, et
al. Lentiviral vector integration in the human genome
induces alternative splicing and generates aberrant
transcripts. J Clin Invest. 2012;122: 1653-66.
34. Ronen K, Negre O, Roth S, et al.
Distribution of lentiviral vector integration sites in mice
following therapeutic gene transfer to treat
beta-thalassemia. Mol Ther. 2011;19:1273-86.
35. Kanter J, Walters MC, Hsieh M, et al.
Outcomes for initial patient cohorts with up to 33 months of
follow-up in the Hgb-206 Phase 1 Trial. Blood.
2018;132:1080-80.
36. Thompson AA, Walters MC, Kwiatkowski
JC, et al. Northstar-2: Updated safety and efficacy analysis
of lentiglobin gene therapy in patients with
transfusion-dependent
b-thalassemia
and non-â0/â0 genotypes. Blood. 2019;134:3543-3543.
37. Lal A, et al. Northstar-3: Interim
results from a phase 3 study evaluating lentiglobin gene
therapy in patients with transfusion-dependent
b-thalassemia
and either a
b0
or IVS-I-110 mutation at both alleles of the HBB Gene.
Blood. 2019; 134:815-15.
38. Gilles AF, Averof M. Functional
genetics for all: engineered nucleases, CRISPR and the gene
editing revolution. Evodevo. 2014;5:43.
39. Palpant NJ, Dudzinski D. Zinc finger
nucleases: Looking toward translation. Gene Ther.
2013;20:121-7.
40. Scharenberg AM, Duchateau P, Smith J.
Genome engineering with TAL-effector nucleases and
alternative modular nuclease technologies. Curr Gene Ther.
2013; 13:291-303.
41. Hoban MD, Bauer DE. A genome editing
primer for the hematologist. Blood. 2016; 127:2525-35.
42. Rossin EJ, Wu DM. CRISPR-based gene
editing: A guide for the clinician. Int Ophthalmol Clin.
2017;57:151-64.
43. Komaroff AL. Gene editing using
CRISPR: Why the Excitement? JAMA. 2017;318: 699-700.
44. Dever DP, Porteus MH, The changing
landscape of gene editing in hematopoietic stem cells: A
step towards Cas9 clinical translation. Curr Opin Hematol,
2017;24: 481-88.
45. Bauer DE, Kamran SC, Lessard S, et
al. An erythroid enhancer of BCL11A subject to genetic
variation determines fetal hemoglobin level. Science.
2013;342:253-7.
46. Psatha N, Reik A, Phelps S, et al.
Disruption of the BCL11A erythroid enhancer reactivates
fetal hemoglobin in erythroid cells of patients with
beta-thalassemia major. Mol Ther Methods Clin
Dev.2018;10:313-26.
47. Bauer DE, Orkin SH. Hemoglobin
switching’s surprise: the versatile transcription factor
BCL11A is a master repressor of fetal hemoglobin. Curr Opin
Genet Dev. 2015;33:62-70.
48. Sankaran VG, Xu JNY Orkin SH,
Transcriptional silencing of fetal hemoglobin by BCL11A. Ann
NY Acad Sci. 2010; 1202:64-8.
49. Alhashem YN, Vinjamur DS, Basu M, et
al. Transcription factors KLF1 and KLF2 positively regulate
embryonic and fetal beta-globin genes through direct
promoter binding. J Biol Chem. 2011;286:24819-27.
50. Ikonomi P, Noguchi CT, Miller W, et
al. Levels of GATA-1/GATA-2 transcription factors modulate
expression of embryonic and fetal hemoglobins. Gene.
2000;261:277-87.
51. Traxler EA, Yao Y, Wang YD, et al. A
genome-editing strategy to treat beta-hemoglobinopathies
that recapitulates a mutation associated with a benign
genetic condition. Nat Med. 2016: 22:987-90.
52. Lin MI, Wang J, Tan Y, et al.
Re-creating hereditary persistence of fetal hemoglobin
(HPFH) to treat sickle cell disease (SCD) and
b-thalassemia.
Blood. 2016;128:4708-4708.
53. Zhang XH, Tee LY, Wang XG, Huag QS,
Yang SH. Off-target effects in CRISPR/Cas9-mediated genome
engineering. Mol Ther Nucleic Acids. 2015;4: e264.
54. Lazzarotto CR, Nguyen NT, Tang X, et
al. Defining CRISPR-Cas9 genome-wide nuclease activities
with CIRCLE-seq. Nat Protoc. 2018;13:2615-2642.
55. Cho SW, Kim S, Kim Y, et al. Analysis
of off-target effects of CRISPR/Cas-derived RNA-guided
endonucleases and nickases. Genome Res. 2014;24:132-41.
56. Cradick TJ, Qiu P, Lee CM, Fine EJ,
Bao G, et al. COSMID: A web-based tool for identifying and
validating CRISPR/Cas off-target sites. Mol Ther Nucleic
Acids.2014;3:e214.
57. Kim D, Bae S, Park J, et al.
Digenome-seq: Genome-wide profiling of CRISPR-Cas9
off-target effects in human cells. Nat
Methods. 2015;12:237-43.
58. Tsai SQ, Joung JK. Defining and
improving the genome-wide specificities of CRISPR-Cas9
nucleases. Nat Rev Genet. 2016;17:300-12.
59. Long J, Hoban MD, Cooper AR, et al.
Characterization of gene alterations following editing of
the beta-globin gene locus in hematopoietic stem/progenitor
cells. Mol Ther. 2018;26: 468-79.
60. Corbacioglu S, Chapin J, Chu-Osier N,
et al. Efficacy results with a single dose of autologous
crispr-cas9 modified cd34+ hematopoietic stem and progenitor
cells in transfusion-dependent
b-thalassemia
and sickle cell disease. EHA Meeting Abstract. S280.
61. Marktel S, Scaramuzza, S, Cicalese MP, et al. Intrabone
hematopoietic stem cell gene therapy for adult and pediatric
patients affected by transfusion-dependent
b-thalassemia.
Nat Med.2019;25:234-41.


