|
|
|
Indian Pediatr 2019;56: 549-550 |
 |
Is Molecular Diagnosis Necessary for Children
with Duchenne Muscular Dystrophy?
|
|
Ratna Dua Puri
From the Institute of Medical Genetics and Genomics,
Sir Ganga Ram Hospital, New Delhi, India
Email: [email protected]
|
|
T
he importance of Duchenne muscular dystrophy
(DMD) for the pediatrician is in the fact that it is the commonest
muscular dystrophy presenting in childhood with an incidence of 1 in
3500 live male births, and also due to the emergence of exciting novel
therapies in recent years. Mutations in the DMD gene that codes
for the dystrophin protein are associated with the severe phenotype of
X-linked progressive muscular dystrophy, DMD, or the milder Becker
muscular dystrophy (BMD). Traditionally, diagnosis of DMD was
established by clinical evaluation with or without enzyme immune-histochemistry
for dystrophin.
The DMD gene is the largest gene in humans
spanning 79 exons, and predominantly expresses in the skeletal and
cardiac muscle with a small amount in the brain [1]. In this issue of
Indian Pediatrics, Tallapaka, et al. [2] characterize the
mutation spectrum in patients presenting with a DMD phenotype, and
demonstrate the decreasing role of invasive muscle biopsy with
increasing availability of non-invasive molecular testing. International
comprehensive guidelines for the management and care of patients with
DMD for practitioners recommend identification of the mutation in
patients. Molecular testing is important for confirming the diagnosis
amidst a plethora of phenotypically similar muscular dystrophies with
high creatine kinase (CK), for counseling families about recurrence risk
and prenatal diagnosis, and for carrier detection of at-risk family
members. In our experience, there exist challenges in counseling for
X-linked disorders in India due to the predominantly patriarchal
society. Hence, utmost care is needed while counseling at-risk carrier
females. Though, carrier females are mostly asymptomatic, monitoring for
cardiac complications and management as needed is important.
Mutations in the gene are predominantly deletions
(60-70%) or duplications (5-10%), concentrated in two hot spot regions
between exons 45-55 and 2-10 for deletions and duplications,
respectively [3-5]. Tallapaka, et al. [3] in their study, also
found 73% of mutations concentrated in these two regions. Appropriately,
they opined that Multiplex Ligation-dependent Probe Amplification (MLPA)
remains the first line investigation for diagnosis of DMD as it tests
for deletions and duplications in all 79 exons of the gene compared to
multiplex Polymerase chain reaction (PCR) that does not test all exons
or define the extent of the deletion. Single exon deletions should be
confirmed by an alternative technique [6]. As availability no longer
remains the issue in India, awareness of the appropriate test is
essential. 25%-35% of pathogenic variants in DMD and about 10%-20% of
males with BMD have single nucleotide variations, small deletions and
insertions or splice site changes identified by sequencing [3]. Given
the high cost of Sanger sequencing for this large gene, next generation
techniques remain the most cost-effective test currently for this cohort
of MLPA-negative patients [4]. In the near future, we can expect a
single step test to evaluate all mutations types in DMD gene [6].
The 79 exons of the DMD gene have a high mutation
rate, and in one of three cases the phenotype occurs due to a de novo
mutation that is not inherited [7]. Thus, new cases will arise despite
the best preventive measures in families with a history of DMD.
The utility of a timely molecular diagnosis is
multifold. Phenotype based on an inframe deletion or duplication that
will not disrupt the reading frame allows translation of the mRNA into a
smaller but active protein. As this protein is active, the phenotype is
the milder BMD. The severe phenotype of DMD occurs when the
deleted/duplicated exons or a premature stop codon impacts translation,
and the resulting truncated dystrophin protein is functionally unstable
[6].
Corticosteroids are currently the accepted standard
of care. The past decade has witnessed many advances in definitive
therapies for DMD. Newer personalized therapies are targeted to work on
patients with specific mutation types [8]. Fig. 1
illustrates what is currently available and approved by the Food and
Drugs Administration (FDA) and European Medicines Agency (EMA) for the
treatment of DMD, and therapies that are in the pipeline.
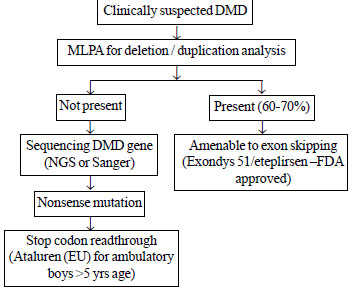
DMD: Duchenne muscular dystrophy; NGS: Next
generation sequencing; MLPA: Multiplex ligation dependant probe
amplification; FDA: Food and Drugs Administration.
|
|
Fig. 1 Precision medicine through molecular diagnosis:
DMD as an example
|
The article by Tallapaka, et al. [2] is very
timely in this era of personalized medicine. Understanding the mutation
spectrum in the population allows development of cost-effective
diagnostic protocols as also define the population potentially amenable
to the existing/developing targeted therapies.
Funding: None; Competing interest: None
stated.
References
1. Falzarano MS, Scotton C, Passarelli C, Ferlini A.
Duchenne muscular dystrophy: From diagnosis to therapy. Molecules.
2015;20:18168-84.
2. Tallapaka K, Ranganath P, Ramachandran A, Uppin
MS, Perala S, Aggarwal S, et al. Molecular and histopathological
characterization of patients presenting with the Duchenne muscular
dystrophy phenotype in a tertiary care center in Southern India. Indian
Pediatr. 2019;56:556-9.
3. Darras BT, Urion DK, Ghosh PS. Dystrophinopathies.
2000 Sep 5. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE
(editors). GeneReviews®. Seattle (WA): University of Washington,
Seattle; 1993-2019. Available from:
https://www.ncbi.nlm.nih.gov/books/NBK1119/. Accessed June 18, 2019.
4. Birnkrant DJ, Bushby K, Bann CM, Apkon SD,
Blackwell A, Brumbaugh D, et al. DMD Care Considerations Working
Group. Diagnosis and management of Duchenne muscular dystrophy, Part 1:
Diagnosis, and neuromuscular, rehabilitation, endocrine, and
gastrointestinal and nutritional management. Lancet Neurol.
2018;17:251-67.
5. Ankala A, Kohn JN, Hegde A, Meka A, Ephrem CL,
Askree SH, et al. Aberrant firing of replication origins
potentially explains intragenic nonrecurrent rearrangements within
genes, including the human DMD gene. Genome Res. 2012;22:25-34.
6. Bello L, Pegoraro E. Genetic diagnosis as a tool
for personalized treatment of Duchenne muscular dystrophy. Acta Myol.
2016;35:122-27.
7. Aartsma-Rus A, Ginjaar IB, Bushby K. The
importance of genetic diagnosis for Duchenne muscular dystrophy. J Med
Genet. 2016;53:145-51.
8. McDonald CM, Campbell C, Torricelli RE, Finkel RS,
Flanigan KM, Goemans N, et al. Clinical Evaluator Training Group;
ACT DMD Study Group. Ataluren in patients with nonsense mutation
Duchenne muscular dystrophy (ACT DMD): A multicentre, randomised,
double-blind, placebo-controlled, phase 3 trial. Lancet.
2017;390:1489-98.
|
|
|
 |
|
