|
|
|
Indian Pediatr 2021;58:30-33 |
 |
Mutation and Phenotypic
Spectrum of Patients With RASopathies
|
|
Meenakshi Lallar, 1 Sunita
Bijarnia-Mahay,1 IC Verma,1
Kaushik Mandal2 and Ratna
Dua Puri1
From 1Institute of Medical Genetics and Genomics, Sir Ganga Ram
Hospital, New Delhi; 2Department of Medical Genetics,
Sanjay Gandhi Postgraduate Institute of Medical Sciences, Lucknow,
India.
Correspondence to: Dr Ratna Dua Puri, Institute of Medical Genetics
and Genomics,
Sir Ganga Ram Hospital, New Delhi, India.
Email: [email protected]
Received: March 30, 2020;
Initial review: April 29, 2020;
Accepted: July 27, 2020.
Published online: August 06, 2020; PII:S097475591600221
|
Objective: To examine the common and specific
clinical features, mutation spectrum and genotype-phenotype correlation
in Noonan syndrome and related RASopathies. Participants: Records
of 30 patients with clinical diagnosis of Noonan syndrome and related
RASopathies presenting over a six-year period at a tertiary care medical
genetics centre were reviewed. Detailed clinical phenotype evaluation
and genetic testing (PTPN11 sequencing or next generation
sequencing) was done. The genetic results were used to classify the
patients. Results: Noonan syndrome was confirmed in 22 patients,
5 had cardiofaciocutaneous syndrome and 3 had Noonan syndrome like
disorder with loose anagen hair. The molecular diagnosis was confirmed
in 27 patients. Mutations in PTPN11 gene were confirmed in 57.8 %
patients. Developmental delay, cardiac defects, ectodermal abnormalities
and coarse face was the predominant phenotype. Noonan syndrome like
disorder with loose anagen hair was clinically identifiable by the
sparse, slow growing hair and caused by one recurrent SHOC2, c.4A>G
mutation. Conclusions: Noonan syndrome and other RASopathies
should be suspected in patients with short stature, cardiac defects,
typical facial dysmorphism with or without ectodermal involvement.
Keywords: Cardio-facio-cutaneous syndrome, Noonan syndrome,
PTPN 11 gene, RAS/MAPK pathway.
|
|
RASopathies are a group of clinically defined
genetic disorders with a prevalence of 1 in 1000. The patients
present with a varying combination of craniofacial, cardiac,
skin and skeletal phenotypes. RASopathies include
neurofibro-matosis type 1 (NF1), Noonan syndrome (NS), Noonan
syndrome with multiple lentigines, Noonan syndrome like disorder
with loose anagen hair (NSLAH), Legius syndrome, Costello
syndrome (CS), cardio-facio-cutaneous syndrome (CFC) and
capillary malformation arteriovenous malformation (CMAVM) [1].
All these disorders have an autosomal dominant pattern of
inheritance with variable expression and penetrance.
In this study, we report on common
phenotypes, diagnostic features, clinical differentiation,
mutation spectrum and genotype – phenotype correlation in
patients with Noonan syndrome and related RASopathies seen over
a six-year period.
METHODS
In this medical record review the clinical
data of patients presenting with Noonan syndrome and related
disorders in our genetic clinic from 2014 through 2019 was
collected on a structured defined proforma. We excluded patients
with neurofibromatosis as they form a distinct, easily
identifiable, group. Informed consent was taken at the time of
evaluation and molecular testing from all patients/parents
included in the study. The PTPN11 gene was
sequenced or next-generation sequencing (NGS) using a
panel/clinical-exome approach on Illumina HiSeq2500 was
performed. All the molecular variants were classified according
to the recommended method of the American College of Medical
Genetics and Genomics [2]. Patients who did not undergo
molecular testing were classified according to the predominant
phenotype. The clinical data is represented as proportions for
frequency of phenotypic features and mutations.
RESULTS
The study cohort included 30 patients, (23
males); 22 of which (16 males) were diagnosed with Noonan
syndrome, five patients with CFC and three patients with NSLAH.
The mean age of patients in the cohort was 7 years [range 4
months to 23 years]. Mutations were identified in 27 patients.
In two patients only PTPN11 sequencing was done, which
was negative and one patient did not consent for molecular
testing.
The age of diagnosis of Noonan syndrome
patients ranged from 4 months to 23 years. The predominant
clinical features were cardiac disease (82%), short stature
(77%), facial dysmorphism (64%), skeletal features like
scoliosis, webbed neck, chest defects (pectus and wide space
nipples) (45%), mild developmental delay (27%), coagulation
abnormalities (23%) and cryptorchidism (14%) (Table I).
The commonest cardiac defect was pulmonary stenosis (39%, 7/18)
followed by hypertrophic cardio-myopathy (33%, 6/18). Skin
features like café au lait macules (size varying from
5-10 mm, more than three) and hyperkeratosis were present in 27%
patients. One child presented at 3 months of age with juvenile
myelo-monocytic leukemia (JMML) syndrome (Table I).
Antenatal features of cystic hygroma, bilateral choroid plexus
cyst and dilated single lymphatic sac were documented in one
child with Noonan syndrome related short stature.
Table I Genotype Phenotype Correlation in 22 Patients with Noonan Syndrome
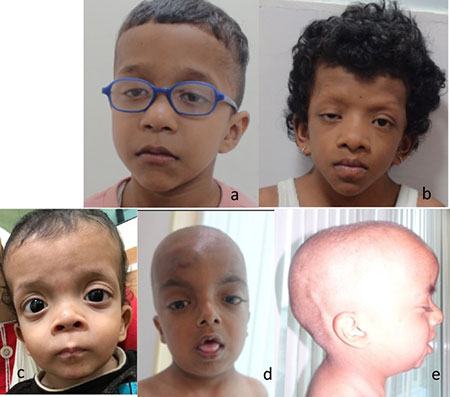
Facial dysmorphism was present in 14 (64%)
patients. (Fig.1 a, b). The four most characteristic
features (hyper-telorism, down-slanting palpebral fissures,
ptosis, and low-set, posteriorly rotated ears) were present
together in only four patients; 10 had atypical facies with one
or two of the above dysmorphic features. Down-slanting palpebral
fissures were seen in 64% and hypertelorism in 57%. Two patients
had coarse facies and ectodermal features and were initially
suspected as CFC syndrome but were later diagnosed as Noonan
syndrome based on genetic testing (RAF1 and SOS2,
respectively) (Fig. 1b).
 |
|
Fig. 1 Variable facial
dysmorphism in Noonan syndrome: (a) Boy with Noonan
Syndrome with hypertelorism, ptosis, downslant palpebral
fissures, low set posteriorly rotated ears (PTPN11, exon
3, c.218C>T), (b) Boy with Noonan syndrome with cardio-facio-cutaneous
syndrome like phenotype – coarse face, woolly hair,
ptosis, hypertelorism, low set posteriorly rotated ears,
(SOS2, Exon 6, c.800T>G), (c) Boy with cardio-facio-cutaneous
syndrome - coarse face, hypertelorism, downslant eyes,
low set ears, coarse hair (BRAF, exon 15, c.1802A>T) and
(d), (e) Boy with Noonan syndrome like disorder with
loose anagen hair - coarse face, hypertelorism,
downslant eyes, relative macrocepahly and the distinct
sparse slow growing hair.
|
Five patients of CFC syndrome were
identified. All had developmental delay, coarse facies and
ectodermal findings (Web Table I) (Fig. 1c).
All the three patients with NSLAH had mild developmental delay,
coarse facies and sparse, slow growing hair (Fig. 1 d, e).
One patient addi-tionally had a history of thrombotic stroke (Web
Table I).
Of the 22 Noonan syndrome patients, mutations
were present in 19 (86%) patients. These were present in
PTPN11 (11/19), SOS1 (2/19), SOS2 (2/19),
RIT 1 (2/19), KRAS (1/19) and RAF1
(1/19) genes. The mutations and related information are listed
in Web Table II. All the identified mutations are
previously reported. The two CFC syndrome patients had the most
common BRAF mutation, c.770A>G, p.Gln257Arg. All three
NSLAH patients harbored the recurrent SHOC2, c.4A>G,
p.Ser2Gly mutation (Web Table I).
DISCUSSION
The clinical diagnosis of Noonan syndrome is
traditionally on a gestalt recognition of the characteristic
facial dysmorphism, cardiac malformations and short stature.
Associated ectodermal features suggest CFC and NSLAH as the
probable diagnosis [3,4]. In this cohort, Noonan syndrome was
the commonest RASopathy (73%), followed by CFC (17%) and NSLAH
(10%). The most consistent and typical facial features in the
Noonan syndrome cohort were down-slanting palpebral fissures,
ptosis and hypertelorism, similar to previous reports [5].
However, we also observed PTPN11 mutation positive Noonan
syndrome with atypical facies, including only hypertelorism,
down-slanting palpebral fissures or ptosis. Another set of
patients with mutations in uncommon Noonan syndrome genes like
RIT1, SOS1 and SOS2 had the typical NS facial
phenotype. A CFC like phenotype was seen with mutations in
RAF1 and SOS2 associated NS suggesting a phenotypic
overlap between NS and CFC. As the facial profile in NS evolves
with age, it alone may be insufficient to predict the genotype,
but along with other systemic features, it can aid in the
clinical diagnosis [6].
The predominant cardiac lesions in NS are
pulmonary stenosis (PS) and hypertrophic cardiomyopathy (HCM).
Early suspicion and echocardiography is important for
appropriate management as PS and HCM in PTPN11 related NS
are seldom rapidly progressive and fatal [7]. Short stature was
another predominant phenotype observed in this study, which may
be due to growth hormone (GH) deficiency, neurosecretory
dysfunction, or GH resistance. GH therapy is approved for Noonan
syndrome and should be initiated early [8].
Renal abnormalities are described in 10-11%
of cases of Noonan syndrome [9]. In the present study one
patient with KRAS associated NS (NS-3) had bilateral
grade 5 vesicoureteric reflux (VUR) with hydronephrosis. VUR
leading to hydronephrosis is previously unreported in Noonan
syndrome. It reiterates the need for multi-organ screening in
malformation syndromes for early detection and management, and
prevention of related morbidity [10]. In one patient (NS-9,
SOS2 mutation) with abnormal gait and brisk deep tendon
reflexes, MRI brain showed bilateral thalamic hyperintensities.
MRI changes in RASopathies are previously reported, but MRI
brain is recommended only if there is abnormality neurological
examination [12]. Bleeding abnormalities are reported in almost
43% patients of NS while on laboratory testing abnormal
coagulation profile is described in upto 90% patients [13]. One
patient with NSLAH had a history of thrombotic stroke. This
previously unreported association is either incidental or a
disease association and needs to be addressed in additional
patient cohorts. Specific PTPN11 gene mutations
predispose to an increased risk of JMML in NS patients [14], but
they have a favorable prognosis and better outcomes,
highlighting the importance of this correlation in management
protocols [15].
A previous Indian study reported exons 3 and
13 of PTPN11 gene as the mutation hot spot in 11 Noonan
syndrome patients [16]. Another study identified exons 3, 8 and
13 of PTPN11 gene with the maximum pathogenic variants in
107 Indian patients [17]. Exons 3, 8, 12 and 13 were the
hotspots exons and the commonest mutation was a previously
reported, c.218C>T in exon 3 in this series. Additionally, the
recurrent SOS2, c.800T>A mutation of NS-9 was also
present in two patients [18]. We observed that most mutations in
Indian patients were similar to those reported in worldwide
literature.
Limitations of this study include a small
number of predominantly NS patients with less representation of
CFC and NSLAH. Also the absence of longitudinal follow up data
limits information on management outcomes and prognosis of the
patients.
Noonan syndrome should be suspected in
patients with short stature (cardiac malformations, primarily
pulmonary stenosis and hypertrophic cardiomyopathy), skeletal
defects and facial dysmorphism (usually includes hypertelorism
and down slanting palpebral fissures). PTPN11 hot spot
exon testing identifies mutations in more than half of Noonan
syndrome patients.
Contributors: ML: study design, article
writing, data collection; ICV: article review, critical input,
study design, data collection; RDP: article critical review and
writing, data collection, study design; SBM: article critical
review, data collection; KM: article critical review, data
collection, PTPN11 test. All authors approved the final version
of manuscript.
Funding; None; Competing interest;
None stated.
| |
|
WHAT THIS STUDY ADDS?
• Most Noonan syndrome patients may
not have all the typical facial gestalt findings, and
Hypertrophic cardiomyopathy is as prevalent as pulmonary
stenosis.
• More than half of Noonan syndrome patients have
mutations in exon 3, 8, 12 and 13 of PTPN11 gene.
|
REFERENCES
1. Tajan M, Paccoud R, Branka S, Edouard T,
Yart A. The Rasopathy family: Consequences of germline
activation of the RAS/MAPK pathway. Endocr Rev. 2018;39:676-700.
2. Richards S, Aziz N, Bale S, et al. ACMG
Laboratory Quality Assurance Committee. Standards and Guidelines
for the Interpretation of Sequence Variants: A Joint Consensus
Recommendation of the American College of Medical Genetics and
Genomics and the Association for Molecular Pathology. Genet Med.
2015;17:405-24.
3. Schulz AL, Albrecht B, Arici C, et al.
Mutation and pheno-typic spectrum in patients with cardio-facio-cutaneous
and Costello syndrome. Clin Genet. 2008;73:62-7
4. Cordeddu V, Di Schiavi E, Pennacchio LA,
et al. Mutation of SHOC2 promotes aberrant protein N-myristoylation
and causes Noonan-like syndrome with loose anagen hair. Nat
Genet. 2009;41:1022-6.
5. Kruszka P, Porras AR, Addissie YA, et al.
Noonan synd-rome in diverse populations. Am J Med Genet A. 2017;
173:2323-34.
6. Allanson JE, Bohring A, Dörr HG, et al.
The face of Noonan syndrome: Does phenotype predict genotype. Am
J Med Genet A. 2010;152A:1960-6.
7. Chen H, Li X, Liu X, et al. Clinical and
mutation profile of pediatric patients with RASopathy-associated
hypertrophic cardiomyopathy: Results from a Chinese cohort.
Orphanet J Rare Dis. 2019;14:29.
8. Seo GH, Yoo HW. Growth hormone therapy in
patients with Noonan syndrome. Ann Pediatr Endocrinol Metab.
2018;23:176-81.
9. George CD, Patton MA, el Sawi M, Sharland
M, Adam EJ. Abdominal ultrasound in Noonan syndrome: A study of
44 patients. Pediatr Radiol. 1993;23:316-8.
10. Golay V, Pandey R, Roychowdhary A.
Chronic tubulo-interstitial nephritis in a solitary kidney of a
child with Noonan syndrome. Indian J Nephrol. 2012;22:304-6.
11. Cizmeci MN, Lequin M, Lichtenbelt KD, et
al. Characteristic MR imaging findings of the neonatal brain in
RASopathies. AJNR Am J Neuroradiol. 2018;39:1146-52.
12. Allanson JE, Roberts AE. Noonan Syndrome.
2001 Nov 15 [Updated 2019 Aug 8]. In: Adam MP, Ardinger
HH, Pagon RA, et al. editors. GeneReviews. University of
Washing-ton, 1993-2020.
13. Nugent DJ, Romano AA, Sabharwal S, Cooper
DL. Eva-luation of bleeding disorders in patients with Noonan
synd-rome: A systematic review. J Blood Med. 2018;9:185-92.
14. Kratz CP, Niemeyer CM, Castleberry RP, et
al. The mutational spectrum of PTPN11 in juvenile
myelomono-cytic leukemia and Noonan syndrome/ myeloproliferative
disease. Blood. 2005;106:2183-5.
15. Jenkins C, Luty SB, Maxson JE, et al.
Synthetic lethality of TNK2 inhibition in PTPN11-mutant leukemia. Sci
Signal. 2018;11(539).
16. Narayanan DL, Pandey H, Moirangthem A, et
al. Hotspots in PTPN11 gene among indian children with Noonan
syndrome. Indian Pediatr. 2017;54:638-43.
17. Athota JP, Bhat M, Nampoothiri S, et al.
Molecular and clinical studies in 107 Noonan syndrome affected
individuals with PTPN11 mutations. BMC Med Genet. 2020;21:50.
18. Yamamoto GL, Aguena M, Gos M, et al.
Rare variants in SOS2 and LZTR1 are associated with Noonan
syndrome. J Med Genet. 2015;52:413-21.
|
|
|
 |
|