|
Case Reports |
||
Indian Pediatrics 2000;37: 93-96 |
||
Partial Lipodystrophy in a Boy |
||
| Ramesh Kumar, Seema, S. Aneja, A.
Seth, V. Taluja From the Department of Pediatrics,
Kalawati Saran Children's Hospital, Lady Hardinge Medical College, New Delhi 110 001,
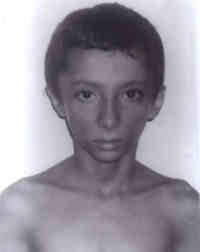
India. Partial lipodystrophy (PL) is a rare condition of unknown etiology, with onset in childhood. It is characterized by progressive loss of subcutaneous fat of face, neck, trunk and upper extremities coupled with C3 hypocomple-mentemia(1), and is four times more common in females than males(2). There are only few reports of partial lipodystrophy in male patients in literature(3,4). Owing to its rarity, we are presenting this case of PL in a boy. Case Report A 9-year-old boy presented with history of progressive loss of fat for the last three years. He was a product of a non-consanguineous marriage, born at term with a birth weight of 3 kg. His development was normal and the child was asymptomatic till 6 years of age when progressive thinning of face was first noted. There was no history of fever, loss of appetite, polyuria, polydypsia or chronic diarrhea. He had been evaluated for tuberculosis and mal-absorption syndromes till it was realized that there was no weight loss but only loss of subcutaneous tissue. There was no history of similar cases in the family. Clinical examination was unremarkable except for a marked sym-metrical atrophy of fat over buccal region and temples (Figs. 1 & 2). His weight, height and mid-arm circumference were 24 kg, 124 cm and 16 cm, respectively. There was no hepato-megaly.
Laboratory evaluation revealed normal levels of hemoglobin, TLC and DLC, serum electrolytes, blood urea, fasting and post-prandial blood sugar, LFT and the thyroid function tests. The lipid profile was also normal with serum cholesterol, high-density lipoprotein, low density lipoprotein and triglyceride levels of 110 mg/dl, 51 mg/dl, 50 mg/dl and 21 mg/dl, respectively. The Mantoux test was negative and urinalysis revealed no abnormality. Plasma C3 level was reduced to 21 mg/dl (Normal 50-120 mg/dl). A diagnosis of partial lipodystrophy was entertained because of painless loss of sub-cutaneous fat over the face associated with C3 hypocomplementemia. Since the patient's main concern was the facial appearance, he was referred to a plastic surgeon. A graft surgery was advised, which was, however, refused by the child's parents. Discussion Partial lipodystrophy (PL) is a rare syn-drome of unknown etiology, first reported by Mitchell in 1885. It has since been described in literature under a variety of titles including progressive lipodystrophy, lipodystrophia progressiva, lipodystrophia facialis, etc. Two main syndromes are included(5): Partial (cephalothoracic) lipodystrophy (Barraquer Simons Disease) and Partial face sparing lipo-dystrophy (Dunningon Kobberling syndrome). Although this entity has been recognized for a long time, the exact etiology and pathogenesis is not well understood. It's association with immunologically related renal disease, systemic sclerosis and high titre of thyroid antibodies supports an immunological basis(5). A neural basis, probably involving the autonomic system is also suggested since experimental studies have shown that the transplanted adipose tissue assumes the characteristics of the area into which it is placed(6). The face sparing variety is inherited, probably as an autosomal dominant trait. There is conflicting data for a possible genetic predisposition for the cephalothoracic type and most cases are sporadic(7). The onset of symptoms is usually between 5-15 years of age and may follow an acute specific fever like measles. The cephalothoracic PL is characterized by slow symmetrical disappearance of facial and subcutaneous fat in upper half of body (the Weir Mitchell type). In some cases, there is co-incidental hypertrophy of subcutaneous fat of lower parts of body (Laignel-Lavastine and Viard type). The face is affected first, and in advanced cases takes on a characteristic cadaverous look with hollow temples, prominant malar bones, chin and zygoma and loss of buccal fat. Eyes may sink deeply into sockets with loss of retro-orbital and periorbital tissue. Smiling produces wrinkles and a prematurely aged expression unshielded by the usual blanket of fat. The veins and muscles of trunk and upper extremities may appear hypertrophied. The overlying skin is of normal color, texture and elasticity. Ten per cent of cases may have hemilipodystrophy involving half of the face or body. The face sparing PL, rarer of the two syndromes, is characterized by loss of subcutaneous fat only from limbs with maintenance of fat on face and trunk or from limbs and trunk with maintenance of facial fat. The progressive atrophy usually ceases after 1.5 to 6 years(3). The histopathology from affected areas reveals either a complete loss or marked decrease in number of fat cells(5,7). PL is usually associated with systemic abnormalities. C3 hypocomplementemia is seen in 70% of patients(7). In a large number of cases, there may be an associated presence of C3 nephritic factor (C3 NeF). A significant number of cases (25-90% according to various reports and reviews)(2,5,8,9) may have renal involvement with biopsy showing mesangio-capillary glomerulonephritis (dense deposit type). Onset of PL and complement abnor- malities antedates the renal disease, sometimes by long periods(9). Many of these patients may have histologically detectable GN before overt clinical manifestations. The proportion of patients who eventually develop significant renal disease is not known. However, in a reported series of 12 patients with PL, 4 died of renal failure after onset of PL(10). These patients may also have abnormal glucose tolerance tests and insulin resistance with overt diabetes in about 20% cases(7). Hypertriglyceridemia is another frequent, but inconsistently reported finding. Other conditions sometimes associated include acanthosis nigricans, hirsuitism, Sjogren syndrome and hyperthyroidism(1). Apart from C3 hypo-complementemia, our patient did not have any other systemic involvement. Thus, PL is not only a cosmetic disability, but may also be a generalized and potentially serious disorder. No specific therapy is avail-able. Patients require close follow up and evaluation for possible complications. Treatments tried with limited success include facial contour restoration with dermal fat grafts, fat injections, microvascular fre-flap grafts and even injections of liquid silicone. New plastic surgical technique using free TRAM flaps has also been employed success-fully(5). Management of GN in patients with PL is same as that of cases without PL. Successful renal transplants in patients with PL and uremia have been reported(11). References 1. Gurbuz O, Yucelten D. Ergun T, Khalilazer R. Partial lipodystrophy. Int J Dermatol 1995; 34: 36-37. 2. Senior B, Gellis SS. The syndromes of total lipodystrophy and of partial lipodystrophy. Pediatrics 1964; 33: 593-612. 3. Poley JR, Stickler GB, Minn R. Progressive lipodystrophy: A clinical study of 50 patients. Am J Dis Child 1963; 106: 356-363. 4. Reardon W, Temple IK, Mackinnon H, Leonard JV, Baraitser M. Partial lipodystrophy syn-dromes_A further male case. Clin Genet 1990; 38: 391-395. 5. Black MM, Cun liffe WJ. Subcutaneous fat. In: Textbook of Dermatology, vol 3, 6th edn. Eds. Champion RH, Burton JL, Burns DA, Breath-nach SM. Oxford, Blackwell Science Ltd., 1998; pp 2403-2436. 6. Langhof H, Zabel R. Zur lipodystrophia pro-gressiva. Arch Klin Exp Dermatol 1960; 210: 313-321. 7. Epstein EH. Lipodystrophy. In: Fitzpatrick's Dermatology in General Medicine, Vol 1, 5th edn. Eds. Freedberg IM, Eisen AZ, Wolff K, Austin KF, Goldsmith LA, Katz SI, et al. NY, McGraw Hill Health Professions Division, 1999; pp 1289-1291. 8. Sissons JGP, West RJ, Fallows J. The complement abnormalities of lipodystrophy. N Engl J Med 1976; 294: 461-465. 9. Bennet WM, Bardana EJ, Wuepper K, Partial lipodystrophy, C3 nephritic factor and clinically inapparent mesangiocapillary glomeruonephritis. Am J Med 1977; 62: 757-760. 10. Simpson NB, Cunliffe WJ, Davison A. Partial lipodystrophy, glomerulonephritis and hypo-complementemia. Br J Dermatol 1979; 101 (Suppl 17): 2429-2430. 11. Ljunghall S, F jellstrom KE, Wibell L. Partial lipodystrophy and chronic hypocomplemen-temic glomerulonephritis. Acta Med Scand 1974; 195: 493-497.
|