Diffuse alveolar hemorrhage (DAH)
refers to accumulation of intra-alveolar red blood cells
originating from alveolar capillaries due to underlying
injury to alveolar microcirculation. The clinical picture
includes hemoptysis, anemia, hypoxemic respiratory failure
with infiltrates on chest X-ray [1,2]. In the absence
of infections or any hemodynamic cause for DAH, anti-neutrophil
cytoplasmic autoantibodies (ANCAs) must be looked for and
ANCA-associated vasculitis (AAV) - mainly microscopic
polyangiitis (MPA),Wegener granulomatosis, or Churg-Strauss
syndrome should be considered. We report a case of MPA who
presented with DAH and was further found to have
interstitial lung disease (ILD).
Case Report
A 13-years-old girl presented with
increasing pallor and gradually worsening dyspnea for last
15 days. She had history of recurrent anemia since the age
of two years, requiring multiple ( four times) blood
transfusions. She had intermittent episodes of cough, fever,
arthralgia, anasarca, and dyspnea on exertion for last 10
years. There was no history of bleeding from any site. She
had received antitubercular therapy (6-months course) one
year back, prior to presentation at our hospital. Her birth,
development and family history was non-contributory.
On examination, her heart rate was
142/min, respiratory rate 48/min, blood pressure 102/60 mmHg
and SpO
2 was 92%
on room air. She had severe pallor, grade-3 clubbing, and
non-blanchable erythematous papular rash over both feet.
Chest examination revealed subcostal and intercostal
retractions with fine crepitations audible all over the
chest. Hepatosplenomegaly was present. Growth was well
preserved. Rest of the systemic examination was normal.
Laboratory Investigations: Blood
investigations revealed anemia (hemoglobin 3.3 g/dL),
platelets 120×10
3
/mm3, total
leukocyte count of 5400/mm3
with normal differential count. Past and present radiographs
showed prominent interstitial reticular shadows in the
middle and lower zones (Fig.1). HRCT chest
showed diffuse ground glass opacities in bilateral lung
fields and interstitial infiltrates with honeycombing
especially in the lower zones (Fig.2). Lung
function tests showed moderate restriction pattern: forced
vital capacity was <64% predicted), and early small airway
obstruction (mean peak expiratory flow rate was <70%
predicted). Echocardiography revealed mild pulmonary artery
hypertension with moderate tricuspid regurgitation. Prussian
blue staining of sputum for hemosiderin-laden macrophages
was positive with siderophage percentage more than 86 %
corresponding to Golde score >100, confirming DAH [1]. Urine
microscopy and renal function tests were normal. ANCA was
positive by immunoflourescence with a perinuclear staining
pattern and ELISA for antimyeloperoxidase ANCA (MPO-ANCA)
was positive (2.808, cutoff-0.394).
 |
|
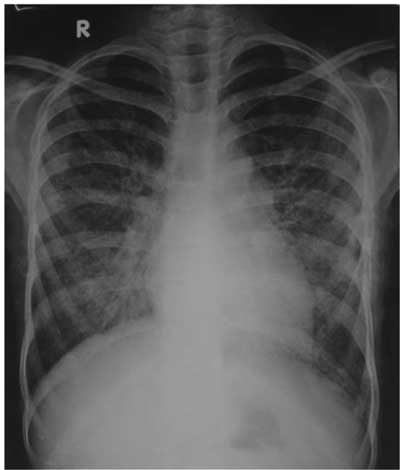
Fig.1 Chest X-ray shows
prominent interstitial reticular shadows due to
diffuse alveolar hemorrhage.
|
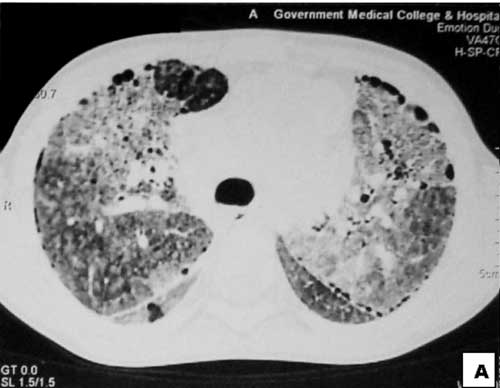 |
|
Fig.2 HRCT of lungs show
bilateral ground glass opacity and interstitial
infiltrates with honeycombing especially in the
periphery.
|
Common infections like malaria, enteric
fever, tuberculosis were ruled out on investigations. Her
ANA and dsDNA were negative. AntiGlomerular Basement
Membrane Antibody and Anti Phospholipid Antibody workup were
negative. Serology for Human Immunodeficiency Virus,
Hepatitis B surface antigen, Hepatitis C and tissue
transglutaminase antibodies were negative. Serum IgE levels
were normal (150IU/mL). Direct Coombs test was negative.
Skin biopsy was unremarkable and did not reveal features of
leucocytoclastic vasculitis.
In view of the multisystem involvement
and positive serology for MPO-ANCA, a diagnosis of MPA was
made. She was started on oral steroids 2 mg/kg/day and
supportive therapy. She went into remission on oral
steroids. After 6 weeks, daily therapy was shifted to
alternate day and then tapered over one year.
Simultaneously, azathioprine was added.
She is on regular follow-up for one year.
She had one relapse in past year which was managed with
short course of steroids. She has remained normotensive,
hepatosplenomegaly has regressed, has had no urinary
complaints or need for blood transfusions (hemoglobin-13.1g/dL),
normal repeat renal functions and, Elisa for MPO-ANCA has
returned to normal.
Discussion
Severe unexplained anemia in a female
with progressive dyspnea and alveolar opacities on chest
imaging, without hemorrhage elsewhere alerted us to the
possibility of DAH. Long history of fever, cough, clubbing,
hepatosplenomegaly, led to the possibility of ILD. Absence
of cutaneous/mucous telangiectasias clinically ruled out
hereditary hemorrhagic telangiectasia. Nonspecific
constitutional symptoms, DAH, ILD with sparing of the upper
airways, no asthma/eosinophilia with positive MPO-ANCA
clinched the diagnosis of MPA.
MPA is a non-granulomatous pauci-immune
primary systemic vasculitides which affects small vessels.
Kidneys and lungs are the most frequently affected organs.
Annual incidence rates of MPA is 2.1-17.5 per million [3].
DAH and ILD have both been reported in MPA [4,5].
However, unlike our case, untreated MPA usually are rapidly
progressive and fatal [4]. Though DAH in MPA is usually
acute, rarely DAH has been reported as part of chronic MPA
in adult patients [6]. The index case had a slow indolent
course with frequent exacerbations which has previously not
been reported in a child. Incidence of ILD is 7.2% in MPA.
Diagnosis of ILD is usually based on radiological evidence
on HRCT and/or lung function tests [5]. Generally, lung
biopsy is not recommended for diagnosis [1,2]. It is
considered if DAH is associated with negative serology and
not a part of a systemic disease. Notably our case didn’t
have renal involvement in past 10 years [1,2].
Two main mechanisms have been proposed
for the development of ILD in patients with small vessel
vasculitis. Firstly, the pulmonary fibrosis occurs in
response to pulmonary haemorrhage and secondly, the ANCA
antigens such as MPO undergo translocation to the surface of
neutrophils (possibly in response to proinflammatory
cytokines), and subsequent binding of circulating ANCA
results in neutrophil degranulation and release of reactive
oxygen species, causing injury and consequent fibrosis
[7,8].
Treatment typically includes
corticosteroids, immunosuppressive agents, and occasionally
plasma-pheresis. 90% of patients achieve remission at 6
months. Relapse rates in AAV are 50%; severe
organ-threatening damage and treatment-related adverse
effects occur in 25% of patients. 10% of those refractory to
standard immunosuppressant therapies are at high risk for
death [9]. Serial hemoglobin measurement guides on DAH
control or progression [1]. Disappearance of ANCA is almost
always associated with absence of disease activity [10].
We conclude that DAH should be considered
as a differential diagnosis in recurrent unexplained anemia.
A high index of suspicion and prompt management can reverse
the symptoms quickly.
Contributors: VM: did case management
& data collection, wrote the draft, interpreted the data;
KR: also helped in management and data collection; SK: did
the radiological reporting; SD: gave intellectual inputs to
case management and edited the manuscript. VM will act as
guarantor of the study. The final manuscript was approved by
all authors.
Funding: None; Competing interests:
None stated.
References
1. Cordier JF, Cottin V. Alveolar
hemorrhage in vasculitis: primary and secondary. Semin
Respir Crit Care Med. 2011;32:310-21.
2. Lara AR, Schwarz MI. Diffuse alveolar
hemorrhage. Chest. 2010;137:1164-71.
3. Gibelin A, Maldini C, Mahr A.
Granulomatosis, microscopic polyangiitis, churg-strauss
syndrome and goodpasture syndrome: vasculitides with
frequent lung involvement. Semin Respir Crit Care Med.
2011;32:264–73.
4. Go´mez-Puerta JA, Herna´ndez-Rodríguez
J, Lo´pez-Soto A, Bosch X. Antineutrophil cytoplasmic
antibody-associated vasculitis and respiratory disease.
Chest. 2009;136:1101-11.
5. Arulkumaran N, Periselneris N, Gaskin
G, Strickland N, Ind PW, Pusey CD, et al.
Interstitial lung disease and ANCA-associated vasculitis: a
retrospective observational cohort study. Rheumatology.
2011;50:2035-43.
6. Martínez-Gabarrón M, Enríquez R,
Sirvent AE, Andrada E, Millán I, Amorós F. Chronic pulmonary
bleeding as the first sign of microscopic polyangiitis
associated with autoimmune thyroiditis. Nefrologia.
2011;31:494-5.
7. Birnbaum J, Danoff S, Askin FB, Stone
JH. Microscopic polyangiitis presenting as a
"pulmonary-muscle" syndrome: is subclinical alveolar
hemorrhage the mechanism of pulmonary fibrosis? Arthritis
Rheum. 2007;56:2065-71.
8. Falk RJ, Terrell RS, Charles LA,
Jennette JC. Anti-neutrophil cytoplasmic autoantibodies
induce neutrophils to degranulate and produce oxygen
radicals in vitro. Proc Natl Acad Sci USA. 1990;87: 4115–9.
9. Langford CA. Treatment of
ANCA-associated vasculitis. N Engl J Med. 2003;349:3-4.
10. Geffriaud-Ricouard C, Noël LH, Chauveau D, Houhou S,
Grünfeld JP, Lesavre P. Clinical spectrum associated with
ANCA of defined antigen specificities in 98 selected
patients. Clin Nephrol. 1993;39:125-36.


