|
|
Case Reports Indian Pediatrics 2001; 38: 194-197 |
|||||||||||||||
|
Chromosomes 6/7 Translocation t(6:7)(q15;32) Presenting as Multiple Pterygium Syndrome |
|||||||||||||||
|
Multiple pterygium (Escobar) syndrome is a rare, autosomal recessive inherited disorder manifested by growth retardation, facial and genital anomalies, and widespread musculo-skeletal deformities. Pterygia-cutaneous webbing usually associated with joint contractures are – the predominant feature of the syndrome(12). In the indexed Indian literature there is only one case report from Turkey of multiple pterygium syndrome in a female child with bilateral optic atrophy(3). In this report we present the clinical, radiographic and laboratory data of a female child morphologically similar to multiple pterygium syndrome with previously unreported features.
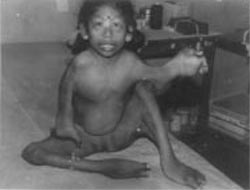
This 5-year-old girl was born to non-consanguineous parents. The mother was 30 years old and the father forty. Two elder and one younger sibling were normal. The girl was born after an unsupervised, uneventful antenatal period by normal vaginal delivery. There was no history of any medication administered during pregnancy. There was no family history of any congenital anomaly. The family belonged to a tribal population inhabiting the hills and forests near Vellore. This child was identified on a peripheral out-reach program and offered medical help. At birth, the child was noticed to have the anomalies with which she presented (see below). Mother observed that her deformities had increased with age. However, no medical intervention had been carried out as the family lived in a remote area with limited access to medical care. On examination, she was a bright co-operative girl, who communicated primarily by gestures. She was unable to walk, but could sit unsupported and could feed herself. Bowel and bladder control was present. Normal hearing was confirmed by an audio-gram. Her weight was 12 kg. The shape of the head was dolichocephalic. She had low-set ears, a high arched palate, absent uvula, a depressed nasal bridge and a narrow chin. Examination of her eyes showed excessive soft tissue folds in both lower eyelids, bilateral absent lacrimal puncta, trichiasis, ulceration of the lower eyelids and conjunctival hyperplasia. Fundus examination was normal. There were multiple pterygia of the neck, axilla, cubital fossae, wrists, and the popliteal region (Fig. 1). Bilateral knee flexion con-tractures and equinus deformities were seen. There also were elbow flexion contractures with bilateral dislocation of the radial heads. Hyperextension of the metacarpophalangeal joints of the index, ring and middle fingers were noted. There was partial syndactyly of all fingers. Cutis laxa and abnormal fat pads in the palms and soles were observed. There was no spine deformity.
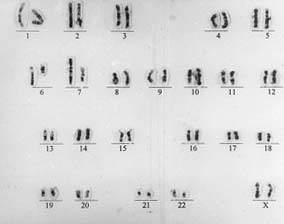
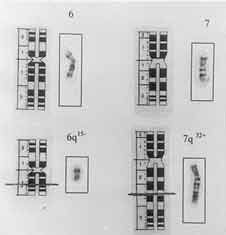
The nipples were widely spaced and were hypoplastic. Labia majora were absent. Labia minora and the introitus were normal. Examination of the cardiovascular system was normal. Chromosomal analysis of the patient using routine GTG Banding technique showed a reciprocal translocation involving chromo-some 6 and 7 (Figs. 2 and 3), described as 46, XX, t(6; 7)(q 15; q 32). The translocation was observed in all the metaphases analyzed. There was no apparent loss of chromosomal material detectable on routine banding technique. The karyotypes of both parents were normal.
The characteristics of this disorder which is rare and mostly transmitted as an autosomal recessive are growth retardation, facial dysmorphia, pterygia resulting in flexion deformities, spine deformities and anomalies of hands, feet and external genitalia. This syndrome must be differentiated from the popliteal pterygium syndrome, which is autosomal dominant with contractures limited to the knee and repercussions that are only functional. It must also be differentiated from arthrogryposis multiplex congenita, which is already complete at birth; and from false secondary pterygia (caudal spinal agenesis, camptomelic syndrome, quadriceps hypo-plasia). The pattern of external malformations in this child closely approximates that of the multiple pterygium (Escobar) syndrome described previously. Dislocation of the radial heads has been described as an uncommon feature earlier(2). It is possible that this may be due to the severity of the contracture. The eye changes have not been previously documented in association with this syndrome. In our case no spine deformity was noticed but this is not a consistent feature of the syndrome(1,2). Escobar syndrome is a recessive disorder and therefore unlikely to be due to a translocation which would simply result in a heterozygous state. There are 3 possible explanations if the translocation is considered to be responsible for an autosomal recessive Escobar syndrome. One explanation is that the patient is a compound homozygote, with a second "molecular mutation" on the homologous chromosome. Secondly, the translocation is acting in a dominant negative fashion, resulting in functional homozygosity. Lastly, the translocation may be entirely coincidental. It is possible that there is an overlap between dominant and recessive pterygia syndromes. If this were a dominant pterygia, then the translocation could explain the phenotype. In all previously reported cases, karyotyping has been normal with the exception of one report of 47XXY/48XXXY mosaicism(4) and one of XY gonadal dysgenesis(5). The reciprocal translocation between chromosomes 6 and 7 seen in our patient has not been reported previously. It appears that the multiple pterygium syndrome in our patient may represent the effect of a chromosomal abnormality which has hitherto not been described. Contributors: VM was the treating orthopedic surgeon and AB was the treating pediatrician of this patient. They were involved in the diagnosis, concept design and drafting of the article and preparation of the manuscript. SD and SK were involved in the chromosomal studies and the analysis of the same. CK and MSS were involved in the critical appraisal of the article and contributed to the intellectural content.
Funding:
None.
|
![]()