|
|
|
Indian Pediatr 2021;58:737-740 |
 |
Clinical
Profile and Outcome of Childhood Autoimmune
Hemolytic Anemia: A Single Center Study
|
|
Kasi Bharathi Thatikonda, Manas Kalra, Arun
Danewa, Pallavi Sachdeva, Tanusree Paul,
Divij Sachdeva, Anupam Sachdeva
From Department of Pediatric Hematology Oncology
and BMT, Institute of Child Health, Sir Ganga Ram
Hospital,
New Delhi.
Correspondence to: Dr Anupam Sachdeva, Director
Pediatric Hematology Oncology and BMT, Institute for
Child Health, Sir Ganga Ram Hospital, New Delhi.
Email:
[email protected]
Received: May 16, 2020;
Initial review: August 01, 2020;
Accepted: December 13, 2020.
Published online: January 2, 2021;
PII: S097475591600296
|
Objective: To analyze
clinical and laboratory parameters, and
treatment outcomes of children with autoimmune
hemolytic anemia (AIHA). Methods:
Retrospective analysis of 50 children aged 0-18
years. Monospecific direct antiglobulin test
(DAT) and investigations for secondary causes
were performed. Disease status was categorized
based on Cerevance criteria. Results:
Median (range) age at diagnosis was 36 (1.5-204)
months. AIHA was categorized as cold
(IgM+,C3+/cold agglutinin+) (35%), warm (IgG+
with/without C3+) (28%), mixed (IgG+, IgM+, C3+)
(15%) and paroxysmal cold hemoglobinuria (4%).
Primary AIHA accounted for 64% cases. Treatment
modalities included steroid (66%), intravenous
immunoglobulin (IVIg) (4%), steroid+IVIg (4%),
and steroid+rituximab (4%). Treatment duration
was longer for secondary AIHA than primary (11
vs 6.6 months, P<0.02) and in patients
needing polytherapy than steroids only (13.3 vs
7.5 months, P<0.006). During median
(range) follow-up period of 73 (1-150) months,
29 (58%) remained in continuous complete
remission, 16 (32%) remained in complete
remission. Conclusion: Infants with AIHA
have a more severe presentation. Monospecific
DAT and a thorough search for an underlying
cause help optimize therapy in most patients of
AIHA.
Keywords: Cerevance criteria, Direct
antiglobulin test, Rituximab, Treatment.
|
|
A utoimmune
hemolytic anemia (AIHA) is caused by the presence of
auto-antibodies directed against antigens on the
surface of red blood cells, leading to premature
destruction [1,2]. AIHA is the main cause of
acquired extra corpuscular hemolysis in children
[1]. AIHA can be subdivided into primary (or
idiopathic) and secondary. AIHA presenting with
thrombocytopenia (Evans synd-rome) tends to have a
more chronic and relapsing clinical course [3-5].
There is scarcity of data on Indian children with
AIHA and their treatment outcome. We present data on
children with AIHA from a single center in India.
METHODS
This is a retrospective analysis
of data from January, 2007 to April, 2019 from our
unit’s database. Fifty children less than 18 years
of age diagnosed with AIHA were enrolled in the
study. AIHA was diagnosed based on the clinical
presentation, positive direct anti globulin test
(DAT) and at least one of the following:
reticulocytosis, haptoglobin <10 mg/dL, and total
bilirubin >1 mg/dL [7]. DAT test was done by gel
card method (Bio-Rad). Infants were evaluated for
TORCH profile. Based on clinical suspicion,
hepatitis B, hepatitis C, HIV serology, Epstein-Barr
virus (EBV) PCR, cytomegalovirus (CMV) PCR,
Mycoplasma pneumoniae antibody test and
antinuclear antibody (ANA) were done. For children
with repeated infections, immunoglobulin profile,
Lymphocyte subset analysis (CD3, CD4, CD8, CD19,
CD16, CD56) was performed. Monospecific DAT test
routinely was performed for all children with
suspected AIHA after the year 2013. AIHA was
categorized based on types of serological antibodies
(IgG, IgM, IgA, C3, and/or combinations) found in
monospecific DAT test, cold agglutinin test and
Donath Landsteiner test. History of drug intake and
recent blood transfusion was obtained for all
patients.
Glucocorticoids were used as the
first line therapy in both warm and cold AIHA. In
cold AIHA, additionally, treatment of the underlying
disease was prioritized. The patient was kept warm
and in cases with very severe anemia, packed red
cell transfusion was given with a heat generator
inside the tubing. Intravenous methyl- prednisolone
(2 mg/kg 8 hourly for 3 days followed by oral
prednisolone, 2mg/kg/day for 4 weeks, then tapered
gradually) was used for patients who were sick,
unable to take orally or had very severe hemolysis.
If there was complete remission
after 4 weeks, tapering of prednisolone was done by
10% with each dose change over a period of 6 months.
If there was no remission/ steroid dependence with
prednisolone dose of 0.2 mg/kg/day, second line
treatment was used. Second-line therapy comprised of
either intravenous immuno-globulin (IVIg), rituximab,
cyclosporine, mycopheno-late mofetil (MMF) or
azathioprine. In steroid dependent cases, one of the
immunosuppressants (cyclosporine, MMF, azathioprine)
was used. In common variable immunodeficiency
(CVID), IVIg was additionally used. Packed red blood
cell transfusion as supportive therapy was given if
the child had a hemoglobin value less than 3 g/dL or
3-6 g/dL with cardiac failure or respiratory
distress and needing intensive care unit (ICU)
care.All patients received folic acid and vitamin
B12 during treatment to support hematopoiesis.
Patients were followed every
month till complete remission and then 3-monthly
till 1 year. Clinical and lab parameters at last
follow-up were classified based on Cerevance
criteria [6] into 4 categories: No remission (NR),
partial remission (PR), complete remission (CR) and
continuous complete remission (CCR).
Statistical analysis: Chi
square test and Student t test (two tailed,
unpaired) were used to compare variables between
primary and secondary AIHA. P value less than
0.05 was considered significant. SPSS version 20.0
was used for the analyses.
RESULTS
Data of 50 children [median
(range) age at diagnosis, 36 months (1.5 months-17
years)] were analyzed. Commonest clinical feature at
diagnosis was pallor (100%) followed by fever (68%)
and jaundice (60%). Hepatomegaly (90%) was seen more
often than splenomegaly (38%).
Mean (SD) hemoglobin at
presentation was 4.7 (1.6) g/dL. Out of 50 children,
72% children presented with very severe anemia
(hemoglobin <3 g/dL, n=6) or severe anemia
(hemoglobin 3-6g/dL n=33, 66%); only 1 child
had mild anemia (>9g/dL). Admission to intensive
care unit (ICU) was needed in 28% children.
Leukocytosis (after correction for nucleated RBCs)
was noted in 27 (54%) patients, and leucopenia in 4
(8%) patients. Only 3 children had thrombocytopenia
at diagnosis. Reticulo-cytosis was seen in 37 cases
(74%), whereas reticulo-cytopenia was seen in 13
cases (26%). Elevated lactate dehydrogenase (LDH)
was seen in 86% children with median (range) LDH
level of 521.5(163- 12858) U/L.
Direct anti-globulin test was 4+
positive in 26 children, 3+ positive in 11 children,
2+ positive in 5 children, and 1+ positive in 5
children.In three DAT-negative children, the
diagnosis was based on clinicopathological suspicion
after ruling out other causes of hemolytic anemia
and on the basis of response to treatment. Two out
of three DAT-negative patients were positive for
Donath Landsteiner test. Monospecific DAT test was
performed in 24 children after its availability from
the year 2013; of which, IgG ± C3 was present in 10
(41%) children, IgM and C3 were present in 3
children (13%) and both IgG and IgM with C3 were
present in 4 children (17%). Cold agglutinin testing
was performed in 21 children and was positive in 13
children. Based on above results cold, warm, mixed
AIHA and PCH was seen in 35%, 28%,15% and 4%
children, respectively. In the other 18% children
seen prior to 2010, AIHA was unclassified.
Secondary AIHA was identified in
36% cases with etiology being infection in 5 (10%) (M.pneumonia,
3; cytomegalovirus infection, 1; Plasmodium vivax
malaria, 1), autoimmune diseases in 5 (10%)
(autoimmune hepatitis, 2; systemic lupus
erythematosus (SLE, 2; and giant cell hepatitis, 1).
Other causes leading to secondary AIHA were Evans
syndrome (6%); childhood malignancies (6%) (Hodgkin
lymphoma, 2 and precursor B cell acute lymphoblastic
leukemia, 1); CVID, 1(2%); and Wiskott Aldrich
syndrome, 1 (2%).
Among infants, hemolysis was
found to be much severe than those who developed
AIHA after infancy (mean (SD) hemoglobin, 3.96
(1.18) vs. 5.13 (1.65) g/dL, P=0.01). In
primary AIHA, the mechanism of hemolysis was more
often IgM and combined antibody mediated than in
children with secondary AIHA wherein it was mainly
IgG-mediated hemolysis.
Steroids alone were used in 33
(66%) children; other medications used were IVIg in
2 children, steroid and IVIg in 2, steroid and
rituximab in 2, steroid, rituximab and cyclosporine
in 1, and steroid and other drugs (three or more) in
7 children. Other immunosuppressive medications used
were mycophenolate mofetil and azathioprine. Among
three patients of Evan syndrome, two patients
responded to first line glucocorticoid therapy and
one responded to second line therapy with IVIg
followed by rituximab. One patient improved
spontaneously and was not given any therapy.
Treatment duration was longer for
children with secondary AIHA than primary (11 vs 6.6
months, P=0.02) and in patients needing
polytherapy than those who improved with steroids
only (13.3 vs 7.5 months, P=0.006). Median
(range) follow up duration was 73 (1-150) months; 29
(58%) remained in CCR, and 16 (32%) in CR (Fig.1).
Despite relapse in 26% cases, 61.5% still showed
good response to steroids. One patient with
secondary AIHA and underlying Hodgkin lymphoma died
due to fulminant fungal sepsis and hemophagocytosis.
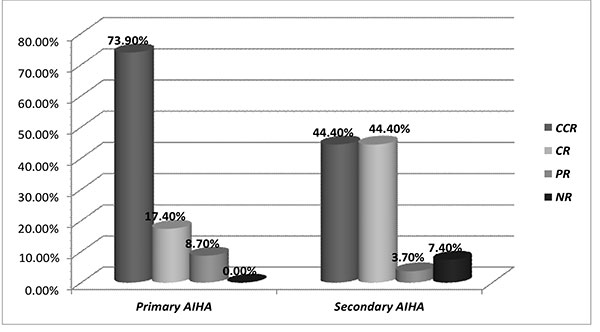
AIHA: autoimmune hemolytic anemia; CCR:
continuous complete remission; CR: complete
remission; PR: partial remission; NR-no
remission.
|
|
Fig. 1 Comparison of remission status
among primary and secondary autoimmune
hemolytic anemia (AIHA).
|
DISCUSSION
We present our institutional data
on pediatric AIHA and underscore the preponderance
of AIHA in younger children; although, the median
age at diagnosis in our study was higher than that
in previous studies (10.8-16 months) [6,7]. Patients
younger than one year required ICU care in view of
severe anemia and hypoxia, similar to the report by
Fan, et al. [7].
In our study, 94% cases had
positive DAT, similar to another Indian study by
Naithani, et al. [8]. Negative DAT in some patients
may be due to low titer of IgG antibodies or IgA or
IgM auto antibody mediated hemolysis.
Reticulocytopenia seen in 26% cases was probably due
to destruction of erythroid progenitors by
autoantibodies [9]. A French national study [6] also
observed a high incidence of reticulocytopenia (39%)
indicating that although reticulocytosis is an
important marker of hemolysis, its absence alone
should not rule out AIHA.
We found that hemolysis was
severe whenever combined or IgM coated antibody
mediated hemolysis occurs. This observation was
similar to previously published study by Sokol, et
al. [10], which showed that compared to IgG mediated
hemolysis alone, IgG along with IgM or IgA leads to
more severe hemolysis. Secondary AIHA was due to
infection in 10% whereas Fan, et al. [7]
showed that infection accounted for 97.6%. In
contrast to this, a French study [6] showed that
secondary forms of AIHA were mainly due to
immunological cause (53%) and infections contributed
to a very small portion (10%). This observation may
be due to increased burden of infection and early
exposure of viruses like EBV in low or middle
in-come countries.
Aladjidi, et al. [6] showed 90%
remission rates with 39% achieving CCR and 51%
attained CR. This may be due to prolonged usage of
steroids (median duration 8 months). Two patients
with PCH had early disease remission. This may be
due to the self-limiting nature of the condition;
however, as per unit policy they were also treated
with short course of steroids. If there was no
remission/steroid dependence with prednisolone in a
dose of 0.2 mg/kg/day, second line treatment was
used [11]. We needed immunosuppressants as second
line of treatment in 26% cases. Rituximab was used
in the standard dose of 375 mg/m 2
per day [12].
Our study is limited by the fact
that it is a retrospective study, comprises of a
small cohort of patients and lacks protocol
uniformity. Treating AIHA in children can be
challenging and may need prolonged and complicated
therapy, especially in secondary AIHA. We suggest
that relapsed or refractory cases of AIHA should be
cared by pediatric hematologists in a tertiary care
center.
Ethics clearance: EC-SGRH;
No.EC/09/20/1715, dated September 30, 2020.
Contributors: KBT: collection
of data, analysis of data, writing of manuscript,
revising it for important intellectual work; PS:
collection of data, analysis of data, writing of
manuscript; TP, DS: collection of data, analysis of
data, writing of manuscript; AD, MK, AS: contributed
patients, final editing of manuscript. All authors
approved the final version of manuscript.
Funding: None; Competing
interest: None stated.
| |
|
WHAT THIS STUDY ADDS?
• Managing AIHA in
infants and those with secondary AIHA is
challenging, with almost one-third patients
at our center needing additional agents to
the steroid backbone.
|
REFERENCES
1. Chou ST, Schreiber AD.
Autoimmune hemolytic anemia. In: Orkin SH,
Fisher DE, Look AT, et al., editors. Nathan and
Oski’s Hematology and Oncology of Infancy and
Childhood. 8th ed. Saunders Elsevier; 2015. p.
411-30.
2. Vaglio S, Arista MC, Perrone
MP, et al. Autoimmune hemolytic anemia in childhood:
Serologic features in 100 cases. Transfusion.
2007;47:50-4.
3. Anderson D, Ali K, Blanchette
V, et al. Guidelines on the use of intravenous
immune globulin for hematologic conditions. Transfus
Med Rev. 2007;21:S9-56.
4. Norton A, Roberts I.
Management of Evans syndrome. Br J Haematol.
2006;132:125-37.
5. Pui CH, Wilimas J, Wang W.
Evans syndrome in childhood. J Pediatr.
1980;97:754-58.
6. Aladjidi N, Leverger G,
Leblanc T, et al. New insights into childhood
autoimmune hemolytic anemia: A French national
observational study of 265 children. Haematologica.
2011;96:655-63.
7. Fan J, He H, Zhao W, et al.
Clinical features and treatment outcomes of
childhood auto immune hemolytic anemia: A
retrospective analysis of 68 cases. J Pediatr
Hematol Oncol. 2016;38:50-55.
8. Naithani R, Agrawal N,
Mahapatra M, Kumar R, Pati HP, Choudhry V.P.
Autoimmune hemolytic anemia in children. Pediatr
Hematol Oncol. 2007;24:309-15.
9. Mangan KF, Besa EC, Shadduck
RK, Tedrow H, Ray PK. Demonstration of two distinct
antibodies in autoimmune hemolytic anemia with
reticulocytopenia and red cell aplasia. Exp Hematol.
1984;12:788-93.
10 Sokol RJ, Hewitt S, Booker DJ,
Bailey A. Red cell autoantibodies, multiple
immunoglobulin classes, and autoimmune hemolysis.
Transfusion. 1990;30:714-17.
11. Ladogana, S, Maruzzi M,
Samperi P, Perrotta S, Vecchio GCD, Farrugggia P.
Diagnosis and Management of Newly Diagnosed
Childhood Autoimmune Haemolytic Anemia.
Recommendations from the Red Cell Study Group of the
Pediatric Haemato-Oncology Italian Association.
Blood Transfus. 2017;15:259-67.
12. Ducassou S, Leverger G,
Fernandes H, Chambost H, Bertrand Y, Armari-Alla C.
Benefits of rituximab as a second-line treatment for
autoimmune haemolyticanaemia in children: A
prospective French cohort study. Br J Haematol.
2017;177:751-58.
|
|
|
 |
|
