|
|
Case Reports Indian Pediatrics 2002; 39:395-398 |
|
Annuloaortic Ectasia in Two Siblings |
|
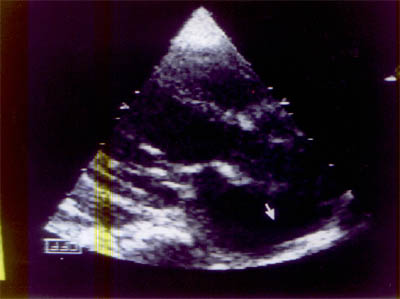
Cardiac lesions in children have diverse manifestations varying from a relatively asymptomatic to an extremely stormy course. We report two brothers aged 4 and 2 years who had minor complaints and were detected to have the rare entity of annuloaortic ectasia (AAE). Case Report The younger sib presented first with cough and breathlessness and was found to have a murmur. Examination revealed bounding pulses, wide pulse pressure, pistol shot sound over the femoral artery and an early diastolic murmur suggestive of aortic regurgitation. A chest radiography showed cardiomegaly with widened mediastinal silhouette. EKG con-firmed left ventricular volume overload. A 2D ECHO clinched the diagnosis of AAE with aneurysmally dilated ascending aorta and arch of aorta with severe aortic regurgitation (Fig. 1). He was put on decongestive drugs. Parents were not in favour of surgery. During the course of the younger siblings investigations, the elder sibling was admitted to the intensive care unit with absent pulses and central cyanosis, probably in a state of cardiogenic shock. He succumbed within an hour of hospitalization. This child had been previously investigated for breathlessness. His X-ray had shown severe cardiomegaly and the 2D ECHO had revealed AAE with aneursymally dilated ascending aorta and the arch of aorta with aortic regurgitation. He had been on decongestive treatment in the form of furosemide, digoxin, enalapril and was partly controlled till the present worsening. A postmortem examination confirmed the same lesions as were seen on the 2D ECHO and in addition showed coarctation distal to the aneurysm: there was no rupture or dissection of the aneurysm. The two siblings had height percentiles within 10th to 25th centiles for the age and did not have any phenotypic features of Marfans syndrome. The parental height was within 50th centile, they were asymptomatic and did not have signs of aortic regurgitation. A 2D ECHO was contemplated but they refused to undertake the study.
Discussion Aneurysm of the aorta, a rare lesion in the pediatric age group refers to pathological dilatation of the aortic lumen involving one or several segments. Thoracic aorta aneurysms are less common than abdominal aorta, are classified by the portion involved; i.e. ascending, arch or descending aorta. The ascending aorta is least commonly affected, both the patients were unique as they had an aneurysm of the ascending and the arch of aorta. Our patients had AAE, a lesion first described by Ellis in 1961(1). The subset of patients had the aneurysm involving the proximal aorta and the annulus causing aortic regurgitation. Between 5 to 10% of the popula-tion undergoing aortic valve replacement for aortic regurgitation are affected by AAE. The lesion is seen more in males than females and the patients present between 4th and 6th decade with severe aortic regurgitation(2). The common histopathological feature with AAE is cystic medial necrosis or degeneration of the affected aortic wall leading to weakening and progressive dilatation. This characteristic lesion was demonstrated in the postmortem histopathological specimen of the ascending aorta of our patient. There was widening of the media due to an increased presence of mucinous ground substance, loss and frag-mentation of elastic fibers. When this process involves the aortic root and the valve, the annulus dilates pulling the leaflets apart and causing regurgitation despite structurally normal leaflets. The association of aortic stenosis and cystic medial necrosis was documented by McKusick in 1957. The condition is also known as Erdheims cystic medial necrosis of aorta. This entity is documented in many families since 1952. There is also evidence for an interrelationship between coarctation, bicuspid aortic valve and abnormal ascending aortic wall(3). Some degree of cystic medial necrosis with AAE is found in all cases of Marfan syndrome(2,4,5). AAE can occur as an isolated entity. In a clinicogenetic study of 18 patients with dilatation of ascending aorta and aortic regurgitation but without evidence of Marfan syndrome except on pathological examination of the aorta, Emanuel et al. reported that 37.3% of 126 first degree relatives had one or more stigmata of Marfan syndrome. Many patients could have the aortic abnormalities of Marfan syndrome without other manifestations. Thus patients with AAE appear to fall into three groups, those with classical Marfan syndrome. those with a forme fruste of the syndrome and those with cystic medial necrosis without any obvious cause seen mainly in adults(6). AAE has been reported with Shprintzen Goldberg syndrome (SGS), which is a generalized connective tissue dysplasia with multiple congenital anomalies(7). Our cases did not have features of either Marfan or SGS syndrome. Molecular genetics permits the character-ization of the protein abnormality and a better understanding of the genotype and phenotype of a disease. The location of the FBNI gene on chromosome 15 has created evidence that there are over 100 mutations in Marfan syndrome, AAE, and other phenotypes such as MASS (mitral valve prolapse, aortic dilation, skin and skeletal manifestations). The abnormality in the FBNI gene and the clinically overlapping conditions is called fibrillinopathies. The FBNI gene encodes the fibrillin protein that is a major structural component of elastin associated microfibrils. The meshwork of elastin and microfibrils in the proximal aorta determines its elasticity and function as an auxiliary pump(5). Around 40% cases of thoracic aorta aneurysms are asymptomatic and the lesion is diagnosed incidentally on routine physical examination or radiography as in our cases. Pulsations over the second and third intercostal spaces, greater intensity of the diastolic murmur to the right of the sternum along with signs of aortic regurgitation can aid the diagnosis. 2D ECHO, CT scan and MRI can confirm the widened and dilated aortic root. Complications include mass effects like superior vena cava syndrome, dyspnea, dys-phagia and hoarseness. Vascular conse-quences namely aortic regurgitation, cardiac failure, myocardial ischemia or infarction, rupture of sinus of valsalva, thromboembolic episodes and dissecting aneurysm often prove fatal. Surgical correction can be achieved with a composite graft comprising of the valve and the conduit. Thus these two cases are unique and represent AAE which was not associated with any syndrome, the presentation was at an earlier age. The etiology is possibly genetic as both sibs were affected and had identical lesions. Acknowledgment We thank Dr. Bharat V. Dalvi, Pediatric Cardiologist, Glenmark Cardiac Center, Mumbai for helping us with his expert opinion on the cases. Contributors: NS collected the clinical data, AK prepared the manuscript, KL did the literature search, BK studied the postmortem specimens. All authors were involved in drafting of the manuscript. KL will act as the guarantor for the manuscript. Funding: None. Competing interests: None stated.
|
| References |
|
![]()