|
|
|
Indian Pediatr 2013;50: 331-333
|
 |
Pseudohypoaldosteronism Type 1: Management
Issues
|
|
Rajni Sharma, Meenu Pandey, Sandeep Kumar Kanwal, and Maria Christina
Zennaro
1,2,3
From the Department of Pediatrics, Division of
Pediatric Intensive Care, Lady Hardinge Medical College and associated
Kalawati Saran Children’s Hospital, New Delhi, India. 1INSERM,
UMRS_970, Paris Cardiovascular Research Center, Paris, France, 2Université
Paris Descartes, Sorbonne Paris Cité, Paris, France, 3Assistance
Publique-Hôpitaux de Paris, Hôpital Européen Georges Pompidou, Paris,
France.
Correspondence to: Dr. Rajni Sharma, Assistant
Professor, Department of Pediatrics, Lady Hardinge Medical College and
associated Kalawati Saran Children’s Hospital, Bangla Sahib Road, New
Delhi 110 001, India.
Email: [email protected]
Received: July 19, 2012;
Initial review: September 06, 2012;
Accepted: September 21, 2012.
|
We report a newborn girl with life-threatening hyperkalemia and salt
wasting crisis due to severe autosomal recessive multiple target organ
dysfunction pseudohypoaldosteronism type 1 (MTOD PHA1). She was
aggressively managed with intravenous fluids, potassium-lowering agents,
high-dose sodium chloride supplementation and peritoneal dialysis.
Genetic analysis revealed a homozygous mutation of the
α- ENaC
(epithelial Na+ channel) gene. She had a stormy clinical course with
refractory hyperkalemia and prolonged hospitalization. Eventually, she
succumbed to pneumonia and septicemia at 4 months of age. This is
probably the first case of PHA1 confirmed by genetic analysis from
India.
Key words: Pseudohypoaldosteronism Type I, Management,
Hyperkalemia.
|
|
Aldosterone is central to fluid and
sodium-potassium balance in the body through its effects in the
distal tubules of the kidney. The hormone acts by binding to the
mineralocorticoid receptor (MR) leading to the induction of
different signaling pathways and transcriptional cascades
modulating the activity of major structural components of ion
transport, including the amiloride-sensitive epithelial Na+
channels (ENaC) [1]. The ENaC is a
heterotrimeric protein complex consisting of
α,
βand
γ
subunits and is widely distributed in other organs as well
including distal colon, salivary and sweat glands [1].
Pseudohypoaldosteronism type 1 (PHA1) is
characterized by end-organ resistance to aldosterone resulting
in salt-losing crisis with hyponatremic dehydration,
hyperkalemia and metabolic acidosis [2]. There are 2 varieties
of PHA-1: the autosomal dominant variety and autosomal
recessive. The former is characterized by NR3C2 gene
mutations affecting the MR and hence resistance is restricted to
the kidney. It has a milder clinical course and generally
resolves spontaneously in childhood. The autosomal recessive
(AR) variety of PHAI results from homozygous or compound
heterozygous mutations in the SCNN1A, SCNN1B or SCNN1G
genes coding for the subunits of ENaC [3].
This is associated with generalized
end-organ resistance or multiple target organ dysfunction (MTOD)
involving a more severe clinical phenotype and requires lifelong
sodium supplementation [2]. We present a newborn with
salt-losing crisis and life-threatening hyperkalemia.
Case Report
A 10-day-old girl was brought to the
emergency department with lethargy and refusal to feed for one
day. There was no history of fever, diarrhea, seizures or
respiratory distress and her urine output was adequate. She was
born at term (birth weight 2.9 kg) to a gravida 2 para 1 mother
with third degree parental consanguinity. The mother had history
of polyhydramnios in the present pregnancy. The 4-year-old elder
male sibling was healthy. On examination, the heart rate was
178/minute, respiratory rate 38/minute, the peripheral pulses
were low volume with a prolonged capillary refill time (4
seconds). Her weight was 2.5 kg at presentation. There was no
evidence of virilization. The rest of the general and physical
examination was normal. Investigations revealed hemoglobin 15.7
g/dL, total leucocyte count 12.1 x 10 9/L,
platelets 2.56 x 109/L,
blood glucose 87 mg/dL, serum sodium 124 mEq/L, serum potassium
10.3 mEq/L, blood urea 84 mg/dL (which normalized later
suggesting prerenal azotemia) and creatinine 0.5 mg/dL. Arterial
blood gas revealed metabolic acidosis (pH 7.15, HCO3 12.7 mmol/L).
Blood and urine cultures were sterile and lumbar puncture was
normal. Chest X-ray and ultrasound abdomen were normal.
Shock was managed with oxygen, fluid boluses and vasopressor
support. Cardioprotective and potassium lowering measures were
immediately started including intravenous calcium gluconate,
sodium bicarbonate, glucose-insulin drip, salbutamol
nebulisation and potassium-exchange resins (sodium polystyrene
sulphonate). She developed ventricular tachycardia and
respiratory failure for which she was intubated and peritoneal
dialysis (PD) was started. The urinary electrolytes revealed
sodium 97 mEq/L and potassium 18 mEq/L. The trans-tubular
potassium gradient (TTKG) at the time of hyperkalemia was 2,
indicating mineralocorticoid resistance. Investigations revealed
high serum aldosterone 2350 pg/ml (normal 10-150 pg/mL) and
direct renin 500 mU/L (normal 2.8-39.9 40 mU/L). Serum
17-alpha-hydroxy progesterone and cortisol levels were normal.
An ultrasound (kidneys, pelviceal system and bladder) including
Doppler scan of the renal vessels and voiding cystourethrogram
were normal. A diagnosis of MTOD PHA1 was considered which was
confirmed by genetic analysis which revealed a homozygous
nonsense mutation c.1339dup on exon 8 of the SCNN1A gene
causing a frameshift in the coding sequence, leading to
substitution of a tyrosine at position 447 by a leucine,
followed by a premature stop codon 12 amino acids downstream
(p.Tyr447Leufs*13) resulting in a truncated
a-ENaC. The
same mutation was found in the heterozygous state in both
parents. Serum electrolytes were within normal limits in the
parents and sibling.
Her general condition improved and the baby
was extubated. Her serum potassium returned to normal and PD was
stopped. She was started on high sodium supplementation 30 mEq/kg
[sodium chloride salt solution 1 g/kg and sodium bicitrate (Shohl’s
solution 15mL/kg)] in divided doses and discharged in a stable
condition on day 37 of life on the same treatment with potassium
binders and mixed feeding (breast milk and formula). She
maintained normal serum electrolytes and gained adequate weight
in weekly follow up visits over the next one month.
The baby was re-admitted at 2 months of age
for pneumonia but no electrolyte imbalance and discharged after
5 days of intravenous antibiotics. Two weeks later, she
presented with lethargy, poor feeding and erythematous papular
rash all over body (Fig. 1). Investigations showed
hyperkalemia (serum potassium 11.4 mEq/L) and hyponatremia
(serum sodium 120 mEq/L) despite oral supplements. She was again
managed with potassium-lowering agents and cardio-protective
measures but hyperkalemia was difficult to control and she
required PD. She failed a trial of high dose fluodrocortisone (upto
1mg). High fluid rates (200 mL/kg/day) were required for
preventing dehydration. Sodium supplementation up to 40 mEq/L
was required to control the hyponatremia initially but later
resulted in hypernatremia (serum sodium 150 mEq/L). Though, the
hyperkalemia improved initially with PD, serum potassium levels
began to rise once it was stopped and PD was repeated on 3 more
occasions. The dose of Kayexalate was increased till tolerated
(6g/kg divided 4 hourly orally and by rectal enema). Thiazide
diuretic (hydrochlorothiazide 2mg/kg) was added and provided
transient relief in the hyperkalemia. She developed nosocomial
pneumonia during the hospital stay and subsequently required
mechanical ventilation. She succumbed to her illness at 4.5
months of age despite aggressive management after more than two
months of ICU stay.
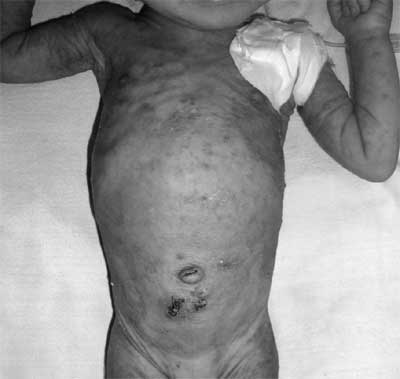 |
|
Fig.1 Erythematous papular rash resembling
Miliaria rubra on the abdomen and arms of the baby. The
photograph was taken after completion of peritoneal
dialysis and sutures can be seen below the umbilicus.
|
Discussion
The presence of hyponatremia and hyperkalemia
in the presence of high aldosterone levels pointed to a
diagnosis of pseudohypoaldosteronism type 1.
Pseudohypo-aldostenism in newborns is known to result
transiently from urinary tract infection, renal dysplasia or
obstructive /reflux nephropathy and should be ruled out as in
our case [4]. The severe clinical presentation suggested
generalized variety or MTOD PHA1 which was confirmed by genetic
analysis of ENCa coding genes.
The erythematous skin rash present in our
patient, which was typically aggravated at the time of
salt-losing crisis, is a characteristic feature of MTOD PHA1 and
results from the blockage and inflammation of exocrine sweat
glands due to high sweat sodium concentration [5]. MTOD PHAI is
also known to be associated with recurrent pneumonia and can
have a clinical presentation similar to cystic fibrosis [6]. The
defective transport of sodium in the airway lumens results in
accumulation of liquid in the airways predisposing to
respiratory disease.
Long-term survival and catch-up growth are
reported in MTOD PHA1 patients treated with NaCl supplementation
and potassium-exchange resins [7]. High doses of sodium (around
10 to 40 mmol/kg NaCl/day) enhance Na +
delivery to the collecting tubules of the kidney and help
increase potassium secretion [8]. Patients require low potassium
diets (0.5 mmol/kg/day) which can be difficult to achieve with
commercial formula milk which contains 15-20 mmol/L of
potassium. Breast milk contains approximately 10 mmol/litre of
potassium and is ideally suited for feeding. It is difficult to
entirely eliminate potassium from the diet as the baby was on
mixed feeding (both breast and formula). High doses of potassium
binders (upto 8g/kg) may be required but are poorly tolerated
orally and may result in rectal bleeding or prolapse when given
as enemas [8]. Infants with MTOD PHAI may require gastrostomy
due to poor oral tolerance of large quantities of fluid, sodium
supplementation and potassium binders [7].
Acute illness can precipitate a salt wasting
crisis in patients with MTOD PHA1. Emergency measures including
PD are sometimes required to control severe hyperkalemia [8]. It
is important to measure serum electrolytes and fluid status
closely during the salt-losing crisis. Indomethacin, a potent
prostaglandin inhibitor, has been observed to reduce the sodium
requirement in patients with MTOD PHA1 though the mechanism of
action is not known [9]. It has no affect on the amount of
potassium-exchange resins required which has led some authors to
use thiazides for the temporary control of hyperkalemia. Though
the use of a diuretic in a salt-losing state may appear
paradoxical, thiazides are postulated to help in urinary
excretion of potassium by increasing the fluid flow to the
distal nephron along with creating electronegativity by
increasing intraluminal chloride [10]. We tried thiazide
diuretics as a last resort to control the dangerous hyperkalemia
in our patient and obtained only transient benefit.
In conclusion, MTOD PHA1 can lead to
salt-losing crisis and life-threatening hyperkalemia in the
neonatal period. Though high-dose sodium supplementation and
potassium-binding resins are the standard of treatment, acute
illness can precipitate severe fluid loss and dangerous
hyperkalemia in these patients which can be particularly
difficult to manage.
Contributors: RS, MP and SKK were
involved in management of the patient. RS and MP reviewed the
literature and drafted the manuscript. SK critically reviewed
the manuscript. MCZ provided the molecular genetic testing for
the proband. All authors approved the final version of the
manuscript.
Funding: None; Competing interests:
None stated.
References
1. Furgeson SB, Linas S. Mechanisms of type I
and type II pseudohypoaldosteronism. J Am Soc Nephrol. 2010;
21:1842-5.
2. Zennaro MC, Hubert EL, Fernandes-Rosa FL.
Aldosterone resistance: structural and functional considerations
and new perspectives. Mol Cell Endocrinol.
2012;350:206-15.
3. Riepe FG. Clinical and molecular features
of type 1 pseudohypoaldosteronism. Horm Res. 2009;72:1-9.
4. Schweiger B, Moriarty MW, Cadnapaphornchai
MA. Severe neonatal hyperkalemia due to pseudohypoaldosteronism
type 1. Curr Opin Pediatr. 2009;21: 269-71.
5. Urbatsch A, Paller AS. Pustular miliaria
rubra: a specific cutaneous finding of type I
pseudohypoaldosteronism. Pediatr Dermatol. 2002;19:317-9.
6. Mora-Lopez F, Bernal-Quiros M,
Lechuga-Sancho AM, Lechuga-Campoy JL, Hernandez-Trujillo N,
Nieto A. Novel mutation in the epithelial sodium channel causing
type I pseudohypoaldosteronism in a patient misdiagnosed with
cystic fibrosis. Eur J Pediatr. 2012 Feb 28. [Epub ahead of
print]
7. Saxena A, Hanukoglu I, Saxena D, Thompson
RJ, Gardiner RM, Hanukoglu A. Novel mutations responsible for
autosomal recessive multisystem pseudohypo-aldosteronism and
sequence variants in epithelial sodium channel alpha-, beta-,
and gamma-subunit genes. J Clin Endocrinol Metab.
2002;87:3344-50.
8. Güran T, Deðirmenci S, Bulut ÝK, Say A,
Riepe FG, Güran Ö. Critical points in the management of
pseudohypoaldosteronism type 1. J Clin Res Pediatr Endocrinol.
2011;3:98-100.
9. Shalev H, Ohali M, Abramson O.
Nephrocalcinosis in pseudohypoaldosteronism and the effect of
indomethacin therapy.J Pediatr. 1994;125:246-8.
10. Stone RC, Vale P, Rosa FC. Effect of
hydrochlorothiazide in pseudohypoaldosteronism with
hypercalciuria and severe hyperkalemia. Pediatr Nephrol.
1996;10:501-3.
|
|
|
 |
|
