|
|
Case Reports Indian Pediatrics 2007;44:228-230 |
||
|
Biotin Responsive Limb Weakness |
||
|
B. Adhisivam
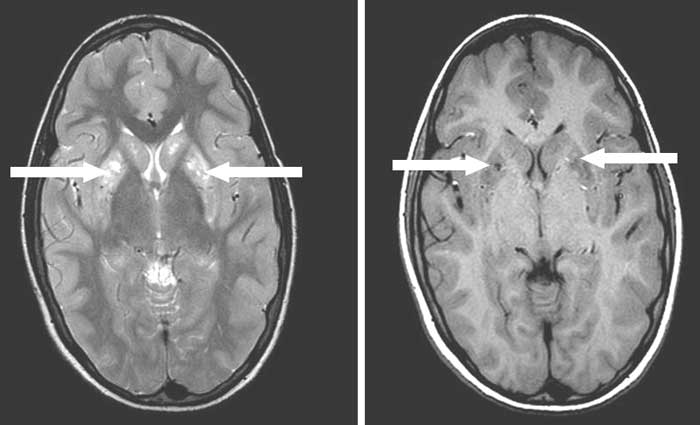
Abstract: Key words: Biotin, Basal ganglia, Egg, Quadriplegia. Acute onset quadriplegia is a frequent neurological problem in children. Guillain Barre syndrome and poliomyelitis present as acute flaccid quadriplegia while injury to head and cervical spine, meningoencephalitis and cerebro-vascular accidents lead to quadriplegia with upper motor neuron signs. Biotin deficiency as a cause of quadriplegia is extremely rare. We describe a 10-year-old boy with biotin responsive basal ganglia disease (BBGD) presenting as spastic quadriplegia. Case Report A developmentally normal and well nourished 10 year old boy was admitted for weakness of all four limbs. Initially, he had frequent falls and an unsteady gait due to weakness of the distal muscles of his lower limbs. Within two weeks, there was rapid progression of the weakness to involve his proximal and trunk muscles so that he could neither move his limbs nor get up from his bed. He was anorexic, lethargic and not conversing. The bladder and bowel functions were normal. There was no history of preceding trauma, fever or seizures. On examination, the child appeared dull and had mask like facies. He had emotional lability and responded only to painful stimuli. The cranial nerves and optic fundii were normal. He had generalized hypotonia for the first two days followed by rigidity. Deep tendon reflexes were exaggerated. Ankle clonus and Babinski’s sign were noted bilaterally. His sensory system was normal. Examination of the other systems including the skin was normal. Investigations revealed the following: Hb 11.8 g/dL, TLC 13,300 × 103/µL, DLC N 55% L42% M1% E2%. The peripheral smear and serum cholesterol were normal. CSF examination was normal and an MRI of his brain showed multiple well defined, symmetric focal lesions in the basal ganglia region with hyperintensity on T2W and hypointensity on T1W sequences (Fig.1).
The basal ganglia lesions were unusual and the etiology for quadriplegia was not clear. Hence, a "Google" search was done to find out the link between the neurodeficit and the radiological findings. Interestingly, a study from Saudi Arabia(1) regarding BBGD matched with the clinico-radiological features of this boy. Hence, biotin deficiency was suspected and a retrospective history revealed that the boy had been consuming raw eggs every alternate day for the past one year for "extra nutrition" which probably predisposed to the deficiency of this vitamin. He was administered oral biotin at a high dose (5 mg/kg/day) and advised not to consume raw eggs. There was remarkable recovery after four days when he started smiling and tried moving his limbs. By the tenth day of therapy, he was able to walk without support and talk normally. He is on daily biotin supplementation and on follow up. Biotin assay and analysis of urine metabolites specific for biotin deficiency could not be done due to financial constraints. Discussion Ozand, et al.(1) described a novel recessive disorder called BBGD (OMIM 607483) characterized by subacute encephalopathy, dysarthria and dysphagia with occasional supranuclear facial nerve palsy or external ophthalmoplegia, and progressing to severe cogwheel rigidity, dystonia and quadriparesis. Brain MRI of these patients revealed specific bilateral necrosis in the head of the caudate nucleus and in the putamen. In view of the similar clinico-radiological picture and therapeutic response to biotin as noted in our case, BBGD was the most probable diagnosis in the 10 year old boy presented here. However, a known predisposing factor (raw egg consumption for one year) cannot be disregarded as coincidental. Probably, the boy had pre-existing marginal biotin deficiency which could have been aggravated by the consumption of raw eggs for a prolonged period. Dietary biotin deficiency produces many symptoms not seen in patients with BBGD, such as dry skin, seborrheic dermatitis, fungal infections and erythematous periorifacial macular rashes(2). Absence of these symptoms in patients with BBGD suggests that sufficient biotin is available in all regions except the brain. The caudate neurodegeneration evident in patients with BBGD suggests that striatal neurons may be particularly susceptible to a lack of adequate biotin. Linkage mapping and positional cloning studies have identified mutations in the transporter gene SLC19A3 as the cause of BBGD(3). The most common basal ganglia disorders with motor symptoms include Wilson’s disease, Hallervorden-Spatz disease, Juvenile Hunting-ton’s disease, L-dopa-responsive dystonia, idiopathic torsion dystonia of childhood, childhood onset parkinsonism and benign acute neurological dysfunction associated with destructive lesions of the basal ganglia(1,4). This biotin-responsive basal ganglia disease is different in many respects from the aforementioned diseases. Other extrapyramidal tract diseases with mitochondrial involvement or Leigh’s subacute encephalomyelopathy usually show either peripheral or CSF lactic acidosis, optic atrophy and other signs of a mitochondriopathy, such as delayed peripheral nerve conduction time. Biotinidase and multiple carboxylase deficiencies have different cortical lesions on MRI(1). The neurological symptoms in this basal ganglia disease disappear within a few days of biotin (5-10 mg/kg/day) administration. However they may reappear within 1 month if biotin is discontinued. Biotin deficiency should be considered in children with acute onset extra pyramidal symptoms and quadriplegia as it can be easily managed without further neurological deterioration. Early molecular diagnosis of BBGD is particularly important to prevent basal ganglia damage, since the progression of the pathology and of the clinical course of symptoms can be prevented simply by the administration of biotin(3). Contributors: BA contributed to case management and drafted the manuscript. DM supervised case management and SM edited manuscript. Funding: None. Competing Interests: None.
| ||
|
References | ||
|
![]()