Acalvaria is a rare congenital anomaly of unknown
pathogenesis in which the flat bones of the cranial vault, duramater and
associated muscles are absent and the facial bones and the base of the
skull are normally present with normal cranial contents(1). To date,
acalvaria has been described as a fatal anomaly(2,3). We report an
infant detected to have acalvaria.
Case Report
A 2-month-old full term boy, delivered normally,
first issue of consanguineous marriage, was referred for an abnormally
soft skull. Prenatal ultrasounds done at 12, 18, 32 and 36 weeks were
reported as normal. There was no history of ingestion of angiotensin
converting enzyme (ACE) inhibitors during pregnancy. The baby was breast
feeding well, had a social smile and normal milestones. On examination
he weighed 5.25 kg and was 58 cm long; head circumference was 40 cm. On
inspection the skull and face appeared normal but on palpation there was
absence of the parietal bones. The facial bones were normal and extended
to the supraorbital ridges; the frontal, temporal and occipital bones
were well felt. The entire bony defect was covered with normal scalp and
skin (Fig. 1). The rest of the physical examination was normal.
An X-ray of the skull showed absence of the parietal bones with
normal facial, frontal, temporal and occipital bones; the infantogram
was normal. A CT scan of the brain showed normal intracranial
structures, no ventricular dilatation and absence of the parietal bones.
The serum calcium was 8.9 mg/dL (9-11 mg/dL), phosphorus was 4.5 mg/dL
(3.5-5.5 mg/dL) and alkaline phosphatase was 265 IU/L (50-350 IU/L).
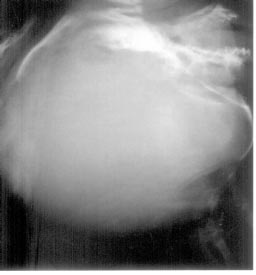 |
|
Fig. 1. Skull with absent parietal bones. |
Discussion
Acalvaria is a rare congenital mal-formation
distinguished by absence of the calvarium. The cranial contents are
usually complete(1), as was seen in our case, though some
neuropathological abnormality is often reported. Hypocalvaria is a
condition in which the cranial bones are hypoplastic(4). Acalvaria may
be associated with holoprosencephaly, hydrocephalus and micropolygyria.
Cardiac anomalies, omphalocele, hypertelorism, cleft lip and palate,
renal tubular dysgenesis, hexadactyly, club foot and congenital
medulloblastoma have been reported(1,5).
The pathogenesis of acalvaria is not exactly known.
Normally during embryo-logical development, after the closure of the
anterior neural pore around the fourth week, migration of the
mesenchymal tissue under the ectoderm underlying the future cerebral
hemispheres takes place. The ectoderm forms the skin and scalp, while
the mesenchyma gives rise to the muscle and bone(6). The most accepted
theory suggests that acalvaria is a postneurulation defect, that is, it
results due to faulty migration of the mesenchyme with normal placement
of the embryonic ectoderm. There is, thus, an absence of the calvarium
but an intact layer of skin over the brain parenchyma(1). Other theories
suggest that it results because of the primary non-closure of the neural
tube or may be a part of a spectrum of anencephaly(7). Acalvaria has
also been described with amniotic bands(8). The disorder is
etiologically and pathogenetically heterogeneous and its prevention by
ingestion of folic acid has not been described. Epidemiological survey
suggest a female predilection. Though one mother has been reported with
two consecutive pregnancies with fetal acalvaria, the condition is not
believed to have a specific risk of recurrence(1). The diagnosis can be
made by the 12th week of gestation by high-resolution transvaginal
ultrasonography(3). The sono-graphic differential diagnosis includes
anencephaly, cephalocele, osteogenesis imperfecta and
hypophosphatasia(1). During pregnancy the alpha-fetoprotein levels are
reported to be very high, while the unconjugated estriol is
undetectable.
Though our patient was born normally by spontaneous
vaginal delivery, pressure on the head during labor and delivery may
cause trauma. To date acalvaria has been described as a fatal anomaly.
Ours is the second reported living case, the first being in Japan(9). As
per our recent communication with the authors, the child reported from
Japan is now 11 year old, and going to a special elementary school for
disabled children. He is severely retarded and disabled.
No surgical procedures to correct the skull defect
have been reported in the newborn period and infancy. Spontaneous bone
growth has been seen in some newborns with scalp defects such as in
cutis aplasia. Conservative management with a careful follow-up and bone
grafting at school age have been recommended(10).
Contributors: VVK and AAN carried out the
clinical workup. AVK, ASK and AAN collected the data and drafted the
manuscript. VVK will act as guarantor of the study.
Funding: HCJMRI, Jehangir Hospital, Pune.
Competing interests: None stated.
