|
Brief Reports |
Indian Pediatrics 2000;37:69-75 |
Chediak-Higashi Syndrome: A Report of Eight Cases From Three Families |
| Mahmoud Al-Sheyyab, Azhar S. Daoud
and Hatem El-Shanti# From the Departments
of Pediatrics and Medical Laboratory Sciences#, Faculty of Medicine, Jordan
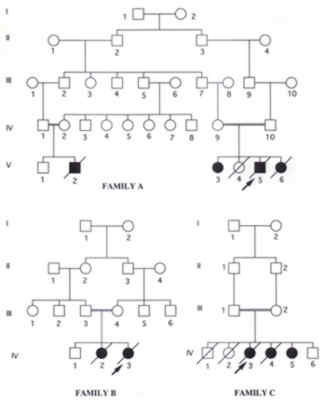
Univeristy of Science and Technology, Irbid, Jordan. Chediak Higashi syndrome (CHS) is a rare autosomal recessive disorder characterized by recurrent pyogenic infections, partial oculocutaneous albinism and abnormally large granules in leukocytes and other granule-containing cells(1). Various features of CHS have been described in small case series and case reports(1-12). In this communication we describe eight cases of CHS from three families with emphasis on their clinical, radiological and some laboratory features, also with a review of the literature. Patients and Methods During a 9 year period (1991-1999) we prospectively collected data on eight patients with CHS from three different families. The diagnosis was based on clinical features and the presence of characteristic giant cytoplasmic granules in the peripheral blood and bone marrow myeloid cells. Relevant clinical data about age, sex, onset, complications and the time and cause of death were collected. A peripheral blood smear and bone marrow were examined by a hematopathologist to confirm the diagnosis. Laboratory tests included blood culture, serum calcium, monospot, Brucella titer, Widal test, bleeding time, prothrombin time (PT), partial thromboplastin time (PTT), hemoglobin electrophoresis and platelet aggregation studies. A skin biopsy from two patients was examined by electron microscopy. Brain CT and MRI were done for one patient who had developmental delay. The brain imaging studies were not done for all patients because of financial reasons. The pertinent details of the three families are summarized below. Family A; Individual V. 4 A male product of a consanguineous marriage was admitted to our hospital several times since the age of 6 months due to recurrent infections. All his vaccinations including BCG were tolerated well. Draining ears with retro-auricular abscesses started at the age of 6 months. He had his first pneumonia at the age of 9 months with right sided lung abscess. Hyperpigmentation of the face and silver-grey hair were noted since the age of 6 months. His growth and development were acceptable till the age of 15 months when his weight and develop-ment started to decline. Anemia, hepatospleno-megaly, clubbing of fingers and toes and petechial rash were noted at the age of 30 months. He died at the age of 4 years with the accelerated phase of the disorder. His family history includes an older sister diagnosed with the same disorder and is still alive at the age of 7 years (V.3). Another sister died at the age of 45 days from presumed sepsis (V.5). Sister V.6 had a similar presentation but died at the age of 30 months. Family B; Individual IV. 3 A 6-month-old female product of consanguineous marriage was admitted due to recurrent infections. She had an older sibling who died at the age of 30 months due to CHS diagnosed and managed elsewhere. Her growth and development were normal. Hyperpigmentation of the face, silver-grey hair and generalized maculopapular rash were noted since the age of 2 months. Three doses of DPT and OPV and hepatitis B vaccine, given at 3, 4 and 5 months of age, as well as, measles vaccine given at 9 months were tolerated well. Chest infections, draining ears and high fever became a problem since then. Anemia and hepatosplenomegaly were noted since the age of 8 months. She died at the age of 30 months with the accelerated phase manifested by massive lymphadenopathy, hepatospleno-megaly and pancytopenia. Family C; Individual IV. 3 An 18-month-old female product of a consanguineous marriage was admitted to our hospital due to repeated infections which started at the age of 4 months. Two siblings, one male and one female had previously died due to repeated infections. Her growth and develop-ment were normal. Hyperpigmentation of the face, silver-grey hair and generalized maculo-papular rash were noted since the age of 4 months. Three doses of DPT and OPV, given at 3, 4 and 5 months of age were tolerated well. Chest infections and draining ears had been a problem since the age of 4 months. Anemia and hepatosplenomegaly were noted since the age of 12 months. By the time of presentation, she had 2 episodes of urinary tract infections, 3 of pneumonia, 3 of otitis media, all presenting with fever. After that she started to have recurrent anemia with hepatosplenomegaly. At that time she was diagnosed to have CHS by bone marrow and peripheral blood film. She died at the age of 35 months with a picture of the acclerated phase of CHS. Individuals IV. 4 and IV. 5 had a similar clinical picture and presenta-tion although the latter is still alive at the age of 6 years. Patient Characteristics Overall, a total of 8 patients, two males and 6 females came from 3 families, two of which (families B and C) belong to the same clan. (Fig. 1).
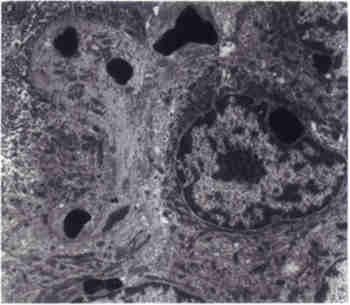
Fig. 1. Pedigrees of the three families. Blackened symbols denote affected individuals diagnosed by both bone marrow examination and blood film examination. All patients had loss of pigment in their irides and retinae with photophobia. Several patients developed nystagmus. One patient showed delayed developmental milestones despite having a normal brain CT and MRI of the brain (Family AV. 4). At the time of presentation all patients had normal PT and PTT and all but one (Family B IV.3) had normal platelet counts. Two patients had prolonged template bleeding time, therefore platelet aggregation studies were carried out (Family AV.4 and Family B IV.3). The former had normal aggregation to ristocetin, thrombin, and collagen but had lower aggregation to ADP. These abnormalities are consistent with storage pool disease. The latter had similar aggregation to control using thrombin, ADP, collagen, and ristocetin at 0.5 and 1.5 mg/ml concentrations. However, this patient had better aggregation when using 0.75 mg/ml ristocetin. Electron microscopy examination of two skin biopsies showed the characteristic electron dense cytoplasmic granules in keratinocytes (Fig. 2).
Fig. 2. Electron microscopy of skin biopsy showing the characteristic electron dense cytoplasmic granules in keratinocyte. One patient (Family C IV.3) had hypo-calcemia with normal serum albumin, phospho-rus and alkaline phosphatase. He also had a Hb F of 8.8% with a normal Hb A2 level. Six patients died due to the accelerated phase. Discussion CHS is an extremely rare disease. Since its initial report, about 200 cases have been reported so far(13). In this communication we describe 8 patients with CHS representing one of the largest series. This means that CHS is not uncommon in Jordan as it is an autosomal reces-sive disorder in a culture where consanguineous marriage is widely practiced(14). The onset of the disease typically starts in infancy with death usually occuring in mid childhood due to overwhelming infections, hemorrhages or the accelerated phase of the disorder. Some patients however, appear to have a clinically milder, "adult" form of the disease(15). The accelerated phase is a diffuse lymphohistiocytic infilterative process associat-ed with adenopathy, organomegaly and pan-cytopenia, seen in about 85% of patients(1,4). All of our patients had their onset in infancy and all experienced repeated infections. The accelerated phase was the terminal event in all of those who died. Besides the abnormalities in the leukocytes, patients with CHS may have giant cytoplasmic granules in the platelets. The platelet count is usually normal except during the accelerated phase. However, patients may have increased bleeding time due to abnormal aggregation caused by storage pool deficiency of ADP and serotonin explaining the bleeding problems in one patient (Family AV.4)(2,4,16,17). Colgan et al. in a study on cats with CHS suggested that the prolonged bleeding time may involve another mechanism in addition to the dense granule deficiency(5). In our study, a storage pool abnormality does not seem to be the sole mechanism for the bleeding diathesis as shown by the increase platelet aggregation with ristocetin using 0.75 mg/ml concentration and normal aggregation with other platelet agonists (Family B IV.3). This defect resembles the platelet aggregation problem seen in von Willebrand disease type II b, or platelet type von Willebrand disease especially with the associated thrombocytopenia present in the same patient. This observation is supported by Ledezma et al. who found defects related to the dense bodies and the alpha granules, as well as platelet membrane abnormalities(7). The bleed-ing diathesis in CHS, therefore appears to be heterogeneous in nature. The outstanding characteristic of CHS is the presence of giant abnormal granules in polymorphonuclear cells, melanocytes, hair, Schwann cells, CNS, peri-pheral ganglia, capillary epithelium, renal tubu-lar epithelium, erythroid precursors, fibroblasts and other granule containing cells(18). Electron microscopic studies on skin biopsies from two of our patients showed abnormal giant cyto-plasmic granules in the keratinocytes. This area needs more explorative efforts. Various neurological abnormalities have been described in CHS(1,3,18,19). The patho-genesis of these abnormalities is either due to lymphohistiocytic infiltration, occuring independent of age or to demylenation/dysmylenation process in patients who live long enough. In our study, only one patient had developmental delay detected early in life with a normal CT/MRI findings, which is probably a coincidental finding. We were unable to do autopsy studies on our patients looking for lymphohistiocytic CNS involvement due to cultural problems. This would be the most likely type of CNS pathology in our patients as it is conforming with their young age. At the cellular level, the excessively large vesicles or granules which are detected in various cell types, are particularly present in cells that are normally engaged in a controlled secretion process such as PMN leukocytes, platelets or cytolytic lymphocytes(1). These granules retain their characteristic ulrastructural and protein composition(20). In non-secreting cell types, such as fibroblasts, the enlarged organelles belong to the late endocytic and lysosomal compartments(21). The gene for CHS has been mapped to chromosome 1q 42-43 by two independent groups and a YAC contig was constructed by the second group(15,22). The CHS gene symbolized LYST for lysosomal trafficking regulator has not been fully cloned as of yet but a 3801 amino acid polypeptide encoded by a 13.5 kb mRNA has been predicted(23). The gene product is believed to participate in the transport machinery into cells(24). A great number of proteins that regulate vesicular transports have been described(25,26). Superfamilies related to the cytoskeleton such as myosin, play a major role in the motility of organelles allowing intracellular transport(27). It is not known whether a defect in any of these proteins or their regulatory factors lead to CHS. Prenatal diagnosis can be performed by finding giant granules or a substantial number of enlarged granules, in fetal blood neurtophils, a technique reported in the feline model of CHS(16). Barrat et al. proposed prenatal diag-nosis at 10-12 weeks gestation in informative females with a small risk of recombination (about 0.5%)(22). One of our patients had hypocalcemia, which has not been reported earlier. There was no apparent cause for the hypocalcemia, although we were unable to do hormonal studies because of financial reasons. This possible association also needs further study. From this series we conclude that bleeding diathesis in CHS is heterogeneous in nature. Skin biopsy is contributory to the diagnosis. Hypocalcemia may be a feature which needs further investiga-tion. This report points to the presence of this disorder amongst Arabs. Acknowledgements The authors are grateful to Dr. D. Todd for reviewing the manuscript and to Mr. M Khdour for performing the Electron Microscopy procedure. References 1. Blume RS, Wolfff SM. The Chediak-Higashi syndrome: Studies in four patients and a review of the literature. Medicine 1972; 51: 247-280. 2. Anderson LL, Paller AS, Malpass D, Schmidt ML, Berger TG. Chediak-Higashi syndrome in a black child. Pediatr Dermatol 1992; 9: 31-36. 3. Ballard R, Tien RD, Nohria, Juel V. The Chediak-Higashi syndrome: CT and MR findings. Pediatr Radiol 1994; 24: 266-267. 4. Barak Y, Nir E. Chediak-Higashi syndrome. Am J Pediatr Hematol Oncol 1987; 9: 42-55. 5. Colgan SP, Hull Thrall MA, Gasper PW. Platelet aggregation and ATP secretion in whole blood of normal cats and cats homozygous and hetero-zygous for Chediak-Higashi syndrome. Blood Cells 1989; 15: 585-595. 6. Kondo N, Shimozawa N, Asano J, Imamura A, Orii T. Chediak-Higashi syndrome with cere-bellar cortical atrophy detected by MRS. Clin Genet 1994; 46: 439-440. 7. Ledezma E, Apitz-Castro R. Protein and glycoprotein abnormalities in platelets from human Chediak-Higashi syndrome: Polyacryl-amide gel electrophoretic study of platelets from five patients. Thromb Res 1985; 40: 19-28. 8. Merino F, Henle W, Ramirez-Duque P. Chronic active Epstein-Barr Virus infection in patients with Chediak-Higashi syndrome. J Clin Immunol 1986; 6: 299-305. 9. Misra VP, King RH, Harding AE, Muddle JR, Thomas PK. Peripheral neuropathy in the Chediak-Higashi syndrome. Acta Neuropathol 1991; 81: 354-358. 10. Price FV, Legro RS, Watt-Morse M, Kaplan SS. Chediak-Higashi syndrome in pregnancy. Obstet Gynecol 1992; 79: 804-806. 11. Tan C, Etcubanas E, Lieberman P, Isenberg H, Murphy ML, King O. Chediak-Higashi syn-drome in a child with Hodgkin's disease. Am J Dis Child 1971; 121: 135-139. 12. Uyama E, Hirano T, Ito K, Nakashima H, Sugimoto M, Naito M, et al. Adult Chediak-Higashi syndrome presenting as parkinsonism and dementia. Acta Neurol Scand 1994; 89: 175-183. 13. King RA, Hearing VJ, Creel DJ, Oetting WS. Albinisim. In: The Metabolic and Molecular Basis of Inherited Disease. Eds. Scriver CR, Baudet A, Sly WS, Valle D. New York, McGraw-Hill, 1995; pp 4353-4392. 14. Al-Salem M, Rawashdeh N. Consanguinity in north Jordan: Prevalence and Pattern. J Biosoc Sci 1993; 20: 553-556. 15. Fukai K, Oh J, Aikarim M, Moore KJ, Kandil HH, Ito H, et al. Homozygosity mapping of the gene for Chediak-Higashi Syndrome to chromo-some 1q42-q44 in a segment of conserved synteny that includes the mouse beige locus (bg). Am J Hum Genet 1996; 59: 620-624. 16. Curnutte JT. Disorders of granulocyte function and granulopoiesis. In: Hematology of Infancy and Childhood, 4th edn. Eds. Nathan DG, Oski FA (eds). Philadelphia, WB Saunders, 1993; pp 916-919. 17. Pratt HL, Carroll RC, Jones JB, Lothrop CD. Platelet aggregation, storage pool deficiency and protein phosphorylation in mice with Chediak-Higashi syndrome. Am J Vet Res 1991; 52: 945-950. 18. Sung JH, Meyers JP, Stadlan EM, Cowan D, Wolf A. Neuropathological changes in Chediak-Higashi disease. J Neuropath Exp Neurol 1969; 28: 86. 19. Maeda K, Sueishi K, Lida M. A Case report of Chediak-Higashi syndrome complicated with systemic amyloidosis and olivo-cerebellar degeneration. Pathol Res Pract 1989; 185: 231-237. 20. Holocombe R, Jones K, Stewart R. Lysosomal enzyme activities in Chediak-Higashi syndrome: Evaluation of lymphoblastoid cell lines and review of the literature. Immunodeficiency 1994; 5: 131-140. 21. Burkhardt JK, Wiebel FA, Hester S, Argon Y. The giant organelles in beige and Chediak-Higashi fibroblasts are derived from late endosomes and mature lysosomes. J Exp Med 1993; 178: 1845-1856. 22. Barrat FJ, Aulgoe L, Pastural E, Largelouse RD, Vilmer E, Cant AJ, et al. Genetic and physical mapping of the Chediak-Higashi syndrome on chromosome 1q42-q43. Am J Hum Genet 1996; 59: 625-632. 23. Karim MA, Nagle DL, Kandil HH, Ruger J, Moore KJ, Spritz RA. Mutations in the Chediak-Higashi syndrome gene (CHS1) indicate requirement for the complet 3801 amino acid CHS protein. Hum Molec Genet 1997; 6: 1087-1089. 24. Baetz K, Isaaz S, Griffiths GM. Loss of cytotoxic T lymphocyte function in Chediak-Higashi syndrome arises from a secretory defect that prevent lytic granule exocytosis. J Immunol 1995; 154: 6122-6131. 25. Rothman JE. Mechanisms of intracellular trans-port. Nature 1994; 372: 55-63. 26. Whiteheart SW, Kubalek EW. SNAPs and NSE: General member of the fusion apparatus. Trends Cell Biol 1995; 5: 64-68. 27. Hirokawa N. Organelle transport along micro-tubules: the role of KIFs. Cell Biol 1996: 6: 135-141. |