|
Case Reports |
Indian Pediatrics 2000;37: 201-203 |
Triploidy Syndrome |
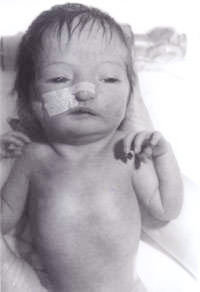
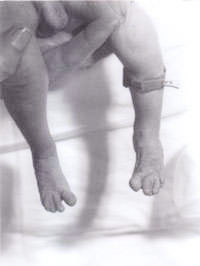
R. Kishore Kumar and Surinder Paul Negi* From the Departments of Pediatrics and *Emergency Medicine, North West Regional Hospital and University of Tasmania, Australia. Reprint requests: Dr. R. Kishore Kumar, Consultant Pediatrician and Lecuturer in Pediatrics and Child Health, North West Regional Hospital and University of Tasmania, P O Box 258, Burnie TAS 7320, Australia. E-mail: [email protected] Manuscript Received: April 5, 1999; Initial review completed: May 26, 1999; Revision Accepted: August 18, 1999 Triploidy syndrome is a rare syndrome and is estimated to occur in about 2 per cent of conceptuses(1). The identification of the syndrome is of utmost importance as most of them die within the first 2 months, and also for the family for further genetic counselling, planning for future family and for their grieving. We report a case to highlight these issues. Case Report A newborn baby was transferred to our hospital from a peripheral center for further assessment because of the "unusual appear-ance". The baby was the second born child to the non-consanguineous Caucasian couple. The mother was aged 30 years. They have a 3-year- old boy, who is healthy and well. The second pregnancy had spontaneously aborted at 14 weeks of gestation. There was no family history of any significant illnesses or any other disorders. The baby was small for gestational age with severe intrauterine growth deficiency with a birth weight of 1600 g at 41 weeks of gestation. He had a large anterior fontanel and a posterior fontanel, low set ears, hypertelorism, pointed nose and a high arched palate. There was syndactaly of third and fourth digits in both upper and lower limbs. Simian crease was noted on both hands. Testes were undescended on both sides and penile length was less than 0.5 cm. Only 2 umbilical vessels were noted. He also had rocker bottom feet on both sides. He also had a small ventricular septal defect confirmed on 2-dimensional echocardiogram. In view of all the above, he was suspected to have trisomy 18. His renal ultrasound scan was reported to be normal. His chromosomal study revealed 18p deletion, but otherwise no evi-dence of any trisomy. In all the cells analyzed, one chromosome 18 homologue appeared atypical within the centrometric region of the short arm. In view of this, parental chromosomal study was performed revealing maternal chromosomes with 18p deletion, exactly same as the baby. But the mother is phenotypically normal with no evidence of dysmorphism and no mental handicap. Hence, this was attributed by the geneticists to be an incidental finding. The baby was susequently reviewed by our senior clinical geneticist, who diagnosed triploidy syndrome and a skin biopsy was performed. The skin fibroblast culture revealed 46XY/69XXY mosaic karyotyping, consistent with the diagnosis of Mosaic Triploidy syndrome. Since our case is a mosaic triploidy, the baby is still alive at the age of 7 months weighing only 3.17 kg with a marked developmental delay. The parents have accepted the reality and have come to terms with it. Since the diagnosis, the number of attendances to casualty for "feeding problems and not gaining weight" has been none, as compared to 10 visits in the first 8 weeks to the general practitioner. The family, being aware of the fact that the risk of recurrence is negligible, are expecting their third child with very little apprehension, though they are planning to have amiocentesis at 16 weeks of gestation to confirm this. The diagnosis of this baby with a triploidy syndrome, made the family come to terms and have enough time to grieve, plan for their next child in a much relaxed manner; helped the general practitioner counsel the family appropriately and the pediatrician to arrange appropriate counselling for their next child. Discussion Triploidy syndrome is characterized by general dysmaturity, muscular hypotonia, large posterior fontanel, low set dysmorphic auricles, hypertelorism, microphthalmia and colobomata, cutaneous syndactaly of third and fourth fingers, simian crease, hypospadias and/or mal-developed external genitalia(2). Most babies are born prematurely and usually live less than one day. There may be evidence of a large placenta with frequent hydatidiform changes. All cases of full triploidy have either been stillborn or have died in the early neonatal period(3), with five months being the longest recorded survival(4). The importance of a correct confirmed diagnosis for any child, in particular for a family who are faced with a "dysmorphic" baby and who are planning to have further children cannot be overemphasized. Syndromal disorders involving trisomies of chromosomes are easily recognized by pediatricians. The question arises when the chromosomes are reported to be normal, when people assume that it is an "idiopathic" problem. Triploidy syndrome is one such syndrome wherein, the triploid cell lines may have disappeared from peripheral blood and the evidence of the triploidy can only be found in the cultured skin fibroblasts. Palliester Killian Syndrome is one other such syndrome where in the tetrasomy 12p has been documented in skin fibroblasts of affected individuals, but not from the peripheral blood. In most instances of triploidy syndrome, the extra set of chromosomes is paternally derived, with 66% attributed to double fertilization, 24% due to fertilization with a diploid sperm and 10% as a fertilization of a diploid egg. Older maternal age has not been a factor unlike most trisomies. There is no data to indicate an increased risk, such as that seen with chromo-somal non-dysjunction which means one can be more optimistic while offering genetic counsell-ing for future children. But in several instances, a triploid pregnancy is either followed or preceded by a molar pregnancy. One has to think of this possibility rather earlier than later, in view of the reduced life span of these babies, so that they can do appropriate tests to offer genetic counselling to the family involved to give them enough time for grieving and come to terms with the reality and also to plan for further pregnancies without being apprehensive. A literature search revealed only 17 citations to this topic in the world so far with the longest survival being 5 months. We report this case to highlight its existence and the prolonged survival of our case probably because of mosaicism.
Fig. 1. Baby with triploidy syndrome.
Fig. 2. Syndactaly of third and fourth toes. Acknowledgements The authors would like to thank Dr. Agnes Bankier, Senior Clinical Geneticist and Dr. Stephen Robertson, Fellow in Clinical Genetics at Royal Children's Hospital, Melbourne for their help in diagnosis and genetic counselling of the family. References 1. Jones KL. Triploidy syndrome and diploid/triploid mixoploidy syndrome. In: Smith's Recognizable Patterns of Human Malformation, 4th edn. Philadelphia, 1988; pp 32-33. 2. Leisti JT, Raivio KO, Rapola MH, Saksela EJ, Aula PP. The phenotype of human triploidy. Birth Defects Orig Artic Ser 1974; 10: 248-253. 3. Garzena E, Farinasso D, Prandi GM, Vardeu P, Bagna R, Cavo L, et al. Triploidy syndrome: A case report. Minerva Pediatr 1995; 47: 307-311. 4. Arvidsson CG. A boy with complete tripolidy and unusually long survival. Acta Paediatr Scand 1986; 75: 507-510.
|