From the Division of Pediatric
Hematology-Oncology, Advanced Pediatric Center and Department of
Histopathology*, Postgraduate Institute of Medical Education and
Research, Chandigarh 160 012 India.
Correspondence to Dr. Deepak Bansal, Assistant
Professor of Pediatrics, Advanced Pediatric Center, PGIMER,
Chandigarh 160 012, India. E-mail:
[email protected]
Manuscript received: September 12, 2003; Initial
review completed: January 13, 2004; Revision accepted: June 29, 2004.
Abstract:
Transfusion-associated graft-versus-host disease
(TA-GVHD) is a dreaded complication in immuno-compromized hosts. The
diagnosis is often delayed because of lack of awareness and the
non-specific clinical features. More than 90% patients succumb to
refractory infections. The only effective preventive measure is
administration of irradiated blood products, which must be made
available in centers managing immunocompromized patients. We report
three cases and discuss pathophysiology and preventive strategies in
this communication.
Key words: Graft versus host disease, Transfusion.
Graft-versus-host disease (GVHD) is the clinical
syndrome ascribed to the inflammatory reaction mounted by the donor
cells against the host organs(1). It was first described in humans
after marrow transplantation (BMT) in 1959(2). Since then, it has
been described in solid organ transplantation(3), blood
transfusion(4) and maternal-fetal transfer of leukocytes(5).
Transfusion-associated GVHD (TA-GVHD) is a dreadful, albeit
infrequent complication of blood transfusion(6), first reported in
1955(7). Inspite of GVHD being a well defined syndrome, the
diagnosis of TA-GVHD is often delayed because of lack of awareness
and the seemingly non-specific manifestations. The relative rarity
of this syndrome prompted us to share our experience of managing
three children with TA-GVHD.
Case Reports
Case 1: A five-year-old girl, a case of acute
lymphoblastic leukemia (L1) presented with fever and lethargy a
fortnight after receiving the late intensification chemotherapy
consisting of vincristine, daunorubicin, cytarabine, etoposide,
thioguanine and oral dexamethasone over a period of five days (UK
ALL X protocol)(8). She had been in a sustained first remission for
nearly 18 weeks. Examination revealed a febrile child with no
localizing features. Investigations revealed a hemoglobin (Hb) of 95
g/L, a while blood cell (WBC) count of 0.04 109/L and a platelet
count of 8 × 109/L. The very severe leucopenia precluded a
differential count. The serum biochemistry, including liver and
renal function tests, was within normal limits. In accordance with
the protocol for febrile neutropenia, she was started on intravenous
cefotaxime and amikacin. A packet red-cell transfusion was
administered on day 3 of admission for anemia (Hb level of 69 g/L).
The blood culture grew Escherchia coli and Streptococcus pneumoniae
that were sensitive to the antimicrobials being administered. Fever
subsided on day 4 of admission. The child appeared to be recovering
till day 9, when she had high-grade fever. It was followed by an
erythematous maculopapular rash, which was first noticed on the
trunk and spread to involve the palms and soles, with periungual and
auricular erythema. Simultaneously, she had loose stools and was
detected to be icteric. Repeat investigations revealed a Hb of 82
g/L, a WBC count of 0.1× 109/L and a platelet count of 11× 109/L.
The serum bilirubin level was 8.4 mg/dL, with a conjugated fraction
of 7 mg/dL. Serum transaminases and alkaline phosphatase levels were
within the normal range. In view of the clinical spectrum of skin
rash, diarrhea, jaundice and fever in an immunocompromised patient,
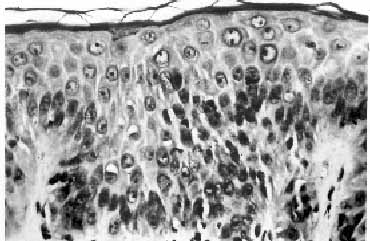
a possibility of TA-GVHD was entertained. A skin biopsy showed focal
vacuolation of basal epithelial cells. There was lymphocytic
infiltration in the stratum malphigi, along with scattered necrotic
keratinocytes. Mild to moderate perivascular infiltration was seen
in the upper dermis. The findings were consistent with GVHD grade II
(Fig. 1). Intravenous infusion of methyl-prednisolone in a dose of
30 mg/kg/day for 3 days was administered. The recovery was dramatic
with subsidence of fever and loose stools within 48 hours. The serum
bilirubin declined progressively to recovery in 12 days. Oral
prednisolone was given for 3 months and then tapered gradually. The
child is in a sustained first remission for nearly a year.
 |
|
Fig. 1. Photomicrograph of skin showing
mild lymphocytic infiltrate with occasional apoptotic bodies
in the epidermis (H&E, 20X). |
Case 2: A six-year-old boy was diagnosed to have
acquired very severe aplastic anemia. He was treated with
antithymocyte globulin (ATG) followed by oral cyclosporine for six
months. He had remained transfusion-free after the administration of
ATG. The while cell counts had recovered. He presented, on an
unscheduled visit, with fever, pallor and gum bleeds. Examination
revealed severe pallor with widespread skin and mucosal bleeds.
Investigations revealed a Hb of 62 g/L, WBC count of 2.5 × 109/L and
a platelet count of 18 × 109/L. Absolute neutrophil count was 0.625
× 109/L. He was transfused two units of packed red cells and a
single unit of platelet concentrate. Febrile neutropenia was managed
with cefotaxime and amikacin, as per standard guidelines. On day 8
of admission, he had high grade fever and a generalized erythematous
maculopapular rash, which involved the palms and soles. He developed
diarrhea two days later. Repeat investigations revealed a persisting
pancytopenia. The total serum bilirubin was 3.5 mg/dL, with a
conjugated fraction of 2.1 mg/dL. Serum transminases and alkaline
phosphatase levels were in the normal range. Blood culture was
sterile. The skin biopsy findings were consistent with a diagnosis
of grade II GVHD. He was administered pulse methylprednisolone
therapy, followed by oral prednisolone. However, his illness
deteriorated progressively and he died a fortnight later, with
complications of infections and bleeds.
Case 3: A four-year-old boy was a diagnosed case
of acquired, very-severe aplastic anemia. Financial constraints
precluded the use of ATG. The blood counts, after six weeks of
monotherapy with cyclosporine A (12 mg/kg/day) were still in the
range designated as very severe(9). During this period, he received
four units of packed red-cells besides multiple units of platelet
concentrates for ongoing bleeding manifestations. A week after the
last red-cell transfusion, he developed high grade fever and a
maculopapular rash on the trunk with progression to all parts of the
body over a period of 48 hours. There were no gastrointestinal
symptoms. The results of investigations were a Hb of 35 g/L; WBC
count of 1.8 109/L and a platelet count of 7 × 109/L. The absolute
neutrophil count was 0.072 × 109/L. Serum bilirubin was 8.4 mg/dL
with a conjugated fraction of 50%. There was no transaminitis; the
serum alkaline phosphatase level was 22 KA units/L (normal 0-13).
The skin biopsy showed features consistent with grade II GVHD. He
was started on pulse methyl-prednisolone. There was no response.
Alpha-hemolytic Streptococcus and Staphylococcus aureus were
isolated from the blood. Antibiotics and antifungals were
administered as per protocol guidelines for the manage- ment of
febrile neutropenia. His condition progressively deteriorated and
culminated in death about three weeks after the onset of GVHD.
Discussion
TA-GVHD is an infrequent complication of blood
transfusion(6). The true incidence of this disorder is not known, as
the manifestations are often mistaken for a viral exanthem or a durg
reaction(6). Two cases of TA-GVHD in neonates, following exchange
transfusion have been reported earlier from our center(10). A
thorough literature search did not yield any other report of TA-GVHD
from India.
Immunologically competent cells in the graft,
transplantation alloantigens in the host and an immunosuppressed
host are the three important prerequisites for the development of
GVHD(6). All cellular blood products contain mature T cells, which
act as immunocompetent cells in the graft and mount GVHD. Routine
blood transfusion are not matched for the major histocompatibility
complex. There are certain alloantigens in the host, which are
lacking in the graft. These foreign antigens stimulate
immunocompetent cells of the graft to produce GVHD. Under normal
circumstances, an immunocompetent host destroys the donor cells,
thereby preventing them to mount a graft-versus-host response.
However, in immunodeficient states, the host cannot reject the
foreign T cells, which proliferate, resulting in GVHD. Rarely TA-GVHD
has been described in an immunocompetent host, when the host and the
graft share HLA haplotype(6). In such a condition, the host does not
recognize the transfused donor cells as foreign and cannot reject
them.
The patients at risk of developing TA-GVHD
include those with (i) congenital immunodeficiencies (ii) acquired
immuno-deficient states (iii) lymphoreticular malignancies (iv)
intensive chemotherapy/radiotherapy and (v) preterm babies and
infants who have received an exchange transfusion(6).
All cellular blood products have been implicated
in TA-GVHD. Transfusions from blood relatives and ‘fresh blood’
components have a large number of viable T cells, thereby enhancing
the risk of TA-GVHD(12).
Pathophysiologically, TA-GVHD is quite similar to
GVHD associated with bone marrow transplantation. However, the
clinical course in TA-GVHD is more fulminant with marrow aplasia and
consequent complication of infections(6). Marrow involvement does
not occur in GVHD associated with BMT, as antigenically, marrow is
similar to the graft. In TA-GVHD, the marrow is antigenically
similar to the host tissue, against which the graft T cells react.
The dominant clinical manifestations in TA-GVHD
are fever and rash as seen in our patients. The median interval
between the transfusion and the onset of fever, which is usually the
first symptom is 10 days, though it can occur as early as 4 days(6).
An erythematous maculopapular rash is observed on the trunk. It then
spreads to involve the extremities, including the palms and soles.
It may progress to generalized erythroderma or bullae formation.
Involvement of the liver is variable. Usually there is mild to
moderate elevation of serum billirubin; predominantly conjugated.
The liver enzymes may be mildly elevated. The serum alkaline
phosphatase is generally raised. Some cases have anorexia, nausea
and occasionally massive diarrhea. Pancytopenia due to bone marrow
aplasia is a late manifestation; occurring after a median interval
of 16 days. Uncontrolled infections are the most common cause of
death which frequently occurs within three weeks of the onset of
GVHD(6). Overall mortality is reported to be more than 90%(6).
A constellation of clinical features related to
skin, gastrointestinal tract, liver and the bone marrow, in an
appropriate setting, must arouse suspicion of TA-GVHD. A lower
threshold for performing skin biopsy aids in supporting the
diagnosis; the findings are however supportive and not pathognomonic.
The histological features in skin biopsy are graded as: (i)
epidermal basal cell vacuolization (grade 1), (ii) mononuclear cell
infiltration and degeneration of epidermal basal layer (grade II),
(iii) bulla formation (grade III), and (iv) ulceration of the skin
(grade IV). Similar findings have been described in drug reactions.
Typical clinical manifestations along with the findings in skin
biopsy are generally sufficient for the diagnosis. Hepatocellular
and cholangiolar cholestasis is associated with degeneration of
small bile ducts. The bone marrow is usually hypocellular with a
lymphocytic or histiocytic infiltration. Demonstration of donor
lympho-cytes in the recipient’s circulation or in the cellular
infiltrates by HLA typing, sex chromatin or DNA analysis is the
diagnostic investigation in an appropriate setting, however is not
routinely available(6).
Mortality in TA-GVHD is high and is attributed to
the complications of acquired bone marrow failure. Various drugs
including high-dose steroids, ATG, cyclosporine, anti-CD3 monoclonal
antibodies, serine protease inhibitors and growth factors have been
tried with no proven benefit(6). There are however, isolated case
reports of successful outcome with one or more of these
drugs(13,14). Pulse methylprednisolone was effective in only one of
our patients. As treatment is largely ineffective, prevention of TA-GVHD
is of paramount importance. Irradiation is the best method to
inhibit proliferation of immunocompetent cells in the donor blood
products(6). Leucocyte depletion and photo-inactivation are the
other not-so-effective methods.
In India, facilities to irradiate blood products
are not available even in tertiary care centers. Directed blood
donations by relative are frequent. These observations suggest that
TA-GVHD is probably more frequent, but under-reported. The features
are often interpreted as sepsis, drug reactions or viral exanthem in
an immunosuppressed host, who is often on multiple drugs. A strong
clinical suspicion in an appropriate setting is required for
clinching the diagnosis. Prevention of TA-GVHD is of utmost
importance; the only effective preventive measure being blood
irradiation. It is imperative that blood irradiation be made
available in medical centers managing immunocompromized patients.
Contributors: AG prepared the initial draft. DB
and RD performed further literature search and critically evaluated
the draft. AD reported the skin biopsy. All contributed in the
review of the manuscript.
Funding: None.
Competing interests: None declared.
