|
|
|
Indian Pediatr 2016;53: 347-348 |
 |
Ghosal Type Hematodiaphyseal Dysplasia
|
|
Amrit Jeevan, #Mathilde
Doyard, Madhulika Kabra, $Valerie
Cormier Daire and Neerja Gupta
From Department of Pediatrics, AIIMS, New Delhi, India; #Imagine
Institut des Maladies Genetiques, France;and $Laboratoire de Genetique
Moleculaire, Institut de Recherche Necker Enfants Malades, Paris,
France.
Correspondence to: Dr Neerja Gupta, Assistant Professor, Division of
Genetics, Department of Pediatrics,
AIIMS, New Delhi, India. Email:
[email protected]
Received: June 24, 2015;
Initial review: September 24, 2015;
Accepted: January 01, 2016.
|
|
Background: Ghosal Type
Hematodiaphyseal Dysplasia is an autosomal recessive disorder
characterized by refractory anemia and diaphyseal bone dysplasia.
Case characteristics: A 3 y 9 mo-old male child presented with
progressive anemia and bowing of thighs. Child was found to have a
previously reported homozygous point mutation c.1238G>A, (p.Arg413Glu)
in Exon 16 of TBXAS1 gene. Outcome: Low dose steroid
therapy resulted in normalization of hemoglobin and prevented further
progression of bony changes. Message: Refractory anemia in
association with bony deformities should prompt pediatricians to
investigate for inherited bony dysplasia.
Keywords: Anemia, Diaphyseal dysplasia,
Engelmann disease.
|
|
Ghosal Type Hematodiaphyseal Dysplasia (GHDD) is a
rare autosomal recessive disorder characterized by predominantly
diaphyseal involvement and corticosteroid sensitive anemia. This entity
was first described by Ghosal in 1988 as a variant of Camurati Engelmann
disease, an autosomal dominant progressive diaphyseal dysplasia [1]. In
2008, David Genevieve, et al. [2] identified mutations in
TBXAS1, a gene at chromosome 7q33-q34 that encodes thromboxane
synthase (TXAS) as the cause of increased bone density in GHDD in four
affected families. There are 24 cases of GHDD reported till date [1-10];
of these 9 are from India. Herein, we report the first case from India
where the diagnosis was confirmed by mutation analysis.
Case Report
A 3 y 9 mo-old boy born to a third degree
consanguineous couple was referred for genetic evaluation for
progressive abdominal distension and pallor since the age of 1 year. The
child first presented to the hospital at 2 years age with severe pallor.
There was no history of bleeding from any site, bony pains, fractures,
visual problems, developmental delay, or seizures. Examination revealed
hepatomegaly (liver span 8 cm) and firm splenomegaly (10 cm below costal
margin). There was no bony deformity, facial dysmorphism, icterus or
lymphadenopathy. Cardio-respiratory and central nervous system
examination were normal. Initial investigations revealed presence of
severe anemia (Hb 46 g/L), and thrombocytopenia (37×10 9/L)
with normal leukocyte count. The reticulocyte count, pre-transfusion
hemoglobin electrophoresis, renal and hepatic functions were normal.
First hour erythrocyte sedimentation rate was 40 mm. Serum iron studies
showed borderline iron deficiency anemia. Bone marrow examination showed
reactive marrow, with prominence of mature lymphoid cells, normoblastic
maturation without any storage/atypical cells. Possible infectious
etiologies were excluded. Enzyme assays for storage disorders were
normal. Fundus evaluation was also normal. Skeletal survey was
unremarkable. He was treated with packed red cell transfusion. By 30
months of age, there was a gradual increase in the blood transfusion
requirement to almost every month. Platelets count ranged from
100-150×109/L. There was no
history of bleeding.
At 3 years 6 months, he developed progressive and
painful limping with deformity of both thighs. His growth parameters at
3 years 9 months were below 5th percentile. Examination showed presence
of bowing of both thighs without any tenderness or fracture. He had
severe pallor. Liver and spleen size were 2.5 cm and 2 cm, respectively.
His hemoglobin and platelets were 56 g/L and 111×10 9/L
respectively without any leucopenia. Radiograph of both femurs showed
evidence of increased cortical density with diaphyseal involvement (Fig.
1). Rest of the bones were normal.
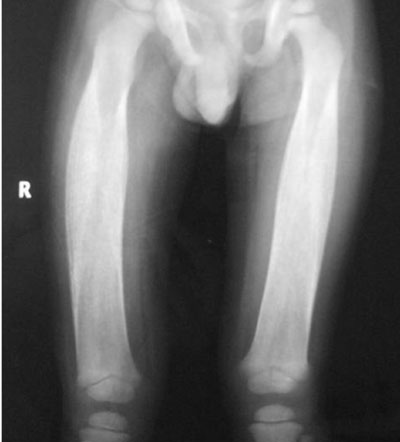 |
|
Fig. 1 Radiograph of both femurs
showing diaphyseal involvement with increased bone density.
|
A diagnosis of Ghosal Type Hematodiaphyseal Dysplasia
was made in view of the involvement of diaphyses of long bones with
refractory anemia. After obtaining an informed consent, mutation
analysis of the TBXAS1 gene was performed by Sanger sequencing of
all the 13 exons and exon-intron boundaries as previously described [2].
It showed presence of a previously reported homozygous mutation
c.1238G>A (p.Arg413Glu) located in exon 16 (NM_001130966.2). Carrier
testing of parents could not be done due to non-availability. The child
was initiated on 1 mg/ kg/ day of oral prednisolone with subsequent rise
of hemoglobin to 88 gm/L after one month of therapy. There was a
decrease in the transfusion requirement. Presently, child is on low dose
prednisolone (0.5 mg/kg/day) and is maintaining his hemoglobin between
80 to 100 g/L.
Discussion
Genevieve, et al. [2] identified the mutations
in TBXAS1, which encodes thromboxane synthase (TXAS). Regulation
of expression of arachidonic acid pathway and genes of tumour necrosis
factor families provides a logical explanation for anemia and increased
bone density associated with this dysplasia. Till date, 24 cases have
been reported worldwide [1-10] (Web Table I). Of these,
total 17 cases are from India and Middle East indicating a common racial
background and probably shared gene pool. Thrombocytopenia was present
only in 50% of the patients. The time lag between hematological symptoms
and bony involvement ranged from 0-60 months with a mean duration 14.5
months. Our case presented with anemia, mild thrombocytopenia and splenomegaly initially followed by diaphyseal involvement of lower
limbs. There was a gap of approximately 2 years for bony changes to
become obvious as the initial skeletal survey was normal initially
resulting in delay in the diagnosis and treatment in our patient.
Four different mutations (c.1463T>C p.Leu488Pro;
c.248T>C p.Leu83Pro; c.1444G>T p.Gly482Trp; c.1238G>A p.Arg413Glu) have
been reported in the 10 cases from 4 different families [2]. Our patient
had c.1238G>A, p.Arg413Glu, mutation which has been reported by this
group in a Pakistani family. This might represent a founder effect and
require mutation analysis of more patients of Indian and Pakistani
origin for further elucidation.
Based upon the underlying molecular basis and
evidence from previous reports, child was initiated on steroid therapy
to which he responded well by maintaining the hemoglobin and cessation
of further progression of bony symptoms. This case illustrates that in
the presence of refractory anemia, thrombocytopenia, hepatosplenomegaly
and bone manifestations, one should further investigate for uncommon
inherited bony dysplasia for timely diagnosis and effective treatment.
Contributors: AJ: reviewed the literature and
wrote the manuscript; NG: Diagnosed and worked up the case and reviewed
the manuscript critically; NG, MK: managed and followed up the case;
VCD, MD: performed molecular analysis.
Funding : None; Competing interests: None
stated.
References
1. Ghosal SP, Mukherjee AK, Mukherjee D, Ghosh AK.
Diaphyseal dysplasia associated with anemia. J Pediatr. 1988;113:49-57.
2. Genevieve D, Proulle V, Isidor B, Bellais S, Serre
V, Djouadi F, et al. Thromboxane synthase mutations in an
increased bone density disorder (Ghosal syndrome). Nat Genet.
2008;40:284-6.
3. Bagga A, Choudhry VP. Diaphyseal dysplasia
[corrected] with anemia and thrombocytopenia. Indian Pediatr.
1989;26:1162-3.
4. Gumruk F, Besim A, Altay C. Ghosal
haemato-diaphyseal dysplasia: A new disorder. Eur J Pediatr.
1993;152:218-21.
5. Alebouyeh M, Vossough P, Tabarrok F. Early
manifestation of Ghosal-type hemato-diaphyseal dysplasia. Pediatr
Hematol Oncol. 2003;20:409-15.
6. Mondal RK, Karmakar B, Chandra PK. Mukherjee K.
Ghosal type hemato-diaphyseal dysplasia: a rare variety of Engelmann’s
disease. Indian J Pediatr. 2007;74:291-3.
7. Mazaheri P, Nadkarni G, Lowe E, Hines P, Vuica M,
Griffin M, et al. Ghosal hematodiaphyseal dysplasia: A rare cause
of a myelophthisic anemia. Pediatr Blood Cancer. 2010;55:1187-90.
8. Vignon SC, Le Merrer M, Vincens A, Monfort M,
Talon P. Ghosal haematodiaphyseal dysplasia: a new case. Arch Pediatr.
2005;12:1244-8.
9. Arora R, Aggarwal S, Deme S. Ghosal
hematodiaphyseal dysplasia – a concise review including an illustrative
patient. Skeletal Radiol. 2015;44:447-50.
10. Datta K, Karmakar M, Hira M, Haldar S, Pramanik
K, Banarjee G. Ghosal hematodiaphyseal dysplasia with myelofibrosis.
Indian J Pediatr. 2013;80:1050-2.
|
|
|
 |
|
